

Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome
- PMID: 19805316
- PMCID: PMC2752572
- DOI: 10.1073/pnas.0905696106
Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome
Abstract
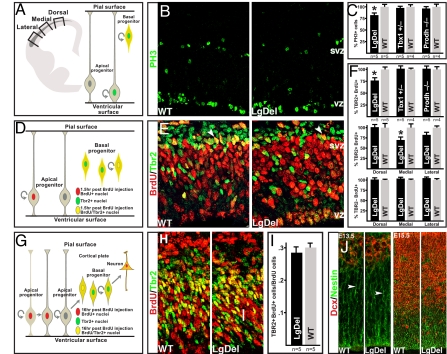
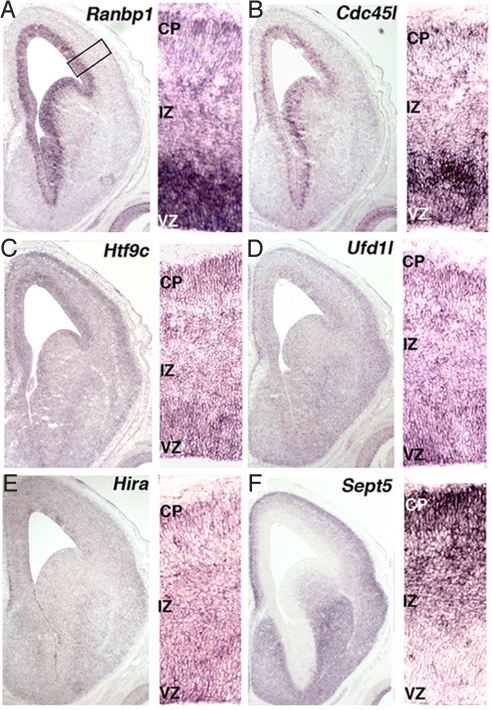
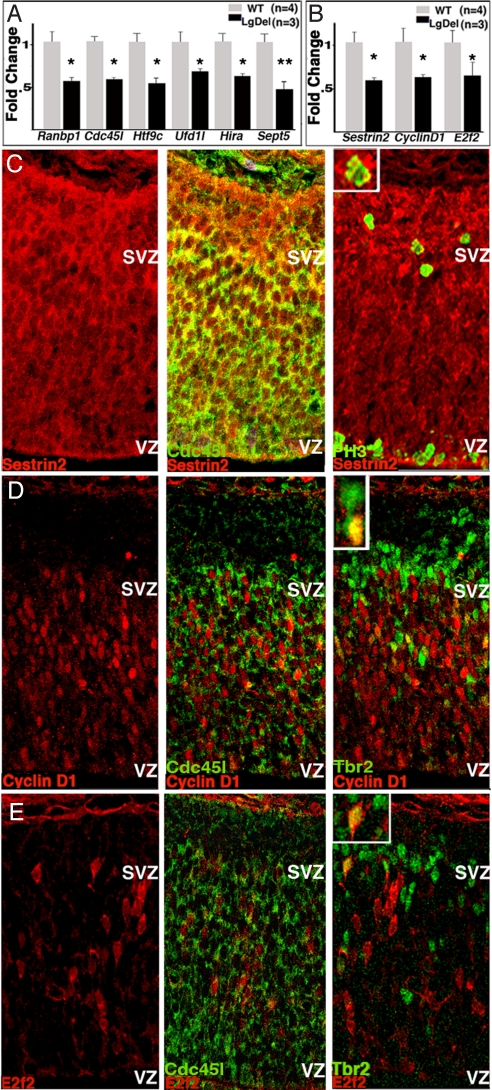
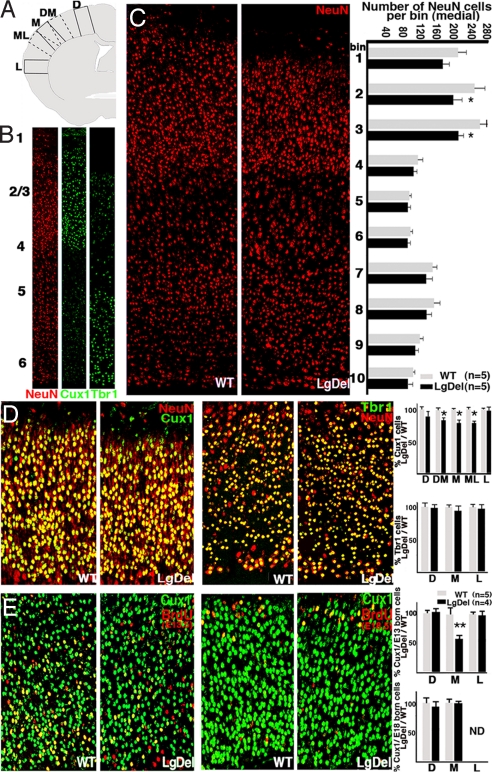
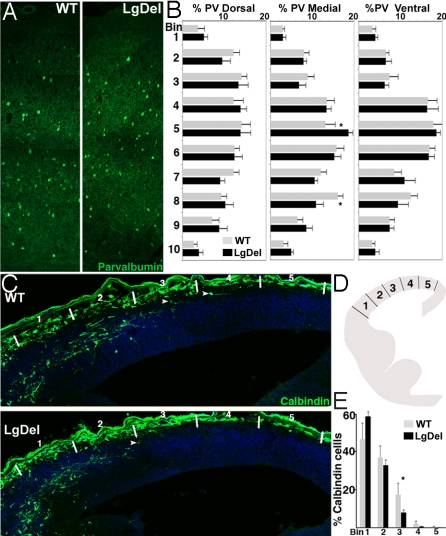
The 22q11 deletion (or DiGeorge) syndrome (22q11DS), the result of a 1.5- to 3-megabase hemizygous deletion on human chromosome 22, results in dramatically increased susceptibility for "diseases of cortical connectivity" thought to arise during development, including schizophrenia and autism. We show that diminished dosage of the genes deleted in the 1.5-megabase 22q11 minimal critical deleted region in a mouse model of 22q11DS specifically compromises neurogenesis and subsequent differentiation in the cerebral cortex. Proliferation of basal, but not apical, progenitors is disrupted, and subsequently, the frequency of layer 2/3, but not layer 5/6, projection neurons is altered. This change is paralleled by aberrant distribution of parvalbumin-labeled interneurons in upper and lower cortical layers. Deletion of Tbx1 or Prodh (22q11 genes independently associated with 22q11DS phenotypes) does not similarly disrupt basal progenitors. However, expression analysis implicates additional 22q11 genes that are selectively expressed in cortical precursors. Thus, diminished 22q11 gene dosage disrupts cortical neurogenesis and interneuron migration. Such developmental disruption may alter cortical circuitry and establish vulnerability for developmental disorders, including schizophrenia and autism.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. - PubMed
-
- Geschwind DH, Levitt P. Autism spectrum disorders: Developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. - PubMed
-
- Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–945. - PubMed
-
- Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res Dev Disabil. 2009;30:763–773. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
