

Inflammatory stress and idiosyncratic hepatotoxicity: hints from animal models
- PMID: 19805476
- PMCID: PMC2763781
- DOI: 10.1124/pr.109.001727
Inflammatory stress and idiosyncratic hepatotoxicity: hints from animal models
Abstract
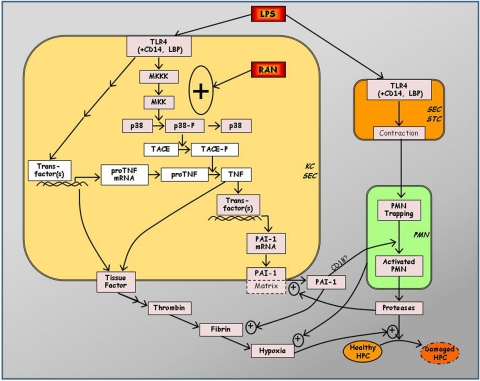
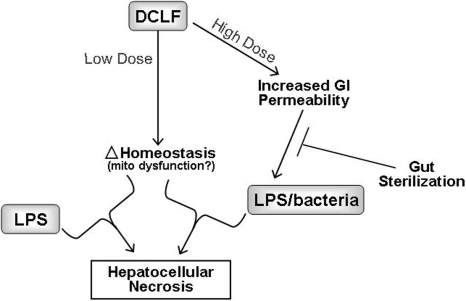
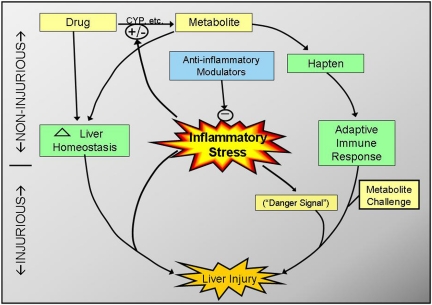
Adverse drug reactions (ADRs) present a serious human health problem. They are major contributors to hospitalization and mortality throughout the world (Lazarou et al., 1998; Pirmohamed et al., 2004). A small fraction (less than 5%) of ADRs can be classified as "idiosyncratic." Idiosyncratic ADRs (IADRs) are caused by drugs with diverse pharmacological effects and occur at various times during drug therapy. Although IADRs affect a number of organs, liver toxicity occurs frequently and is the primary focus of this review. Because of the inconsistency of clinical data and the lack of experimental animal models, how IADRs arise is largely undefined. Generation of toxic drug metabolites and induction of specific immunity are frequently cited as causes of IADRs, but definitive evidence supporting either mechanism is lacking for most drugs. Among the more recent hypotheses for causation of IADRs is that inflammatory stress induced by exogenous or endogenous inflammagens is a susceptibility factor. In this review, we give a brief overview of idiosyncratic hepatotoxicity and the inflammatory response induced by bacterial lipopolysaccharide. We discuss the inflammatory stress hypothesis and use as examples two drugs that have caused IADRs in human patients: ranitidine and diclofenac. The review focuses on experimental animal models that support the inflammatory stress hypothesis and on the mechanisms of hepatotoxic response in these models. The need for design of epidemiological studies and the potential for implementation of inflammation interaction studies in preclinical toxicity screening are also discussed briefly.
Figures
References
-
- Aggarwal BB, Kohr WJ, Hass PE, Moffat B, Spencer SA, Henzel WJ, Bringman TS, Nedwin GE, Goeddel DV, Harkins RN. (1985) Human tumor necrosis factor. Production, purification, and characterization. J Biol Chem .260: 2345–2354 - PubMed
-
- Aithal GP, Day CP. (2007) Nonsteroidal anti-inflammatory drug-induced hepatotoxicity. Clin Liver Dis .11: 563–575, vi–vii - PubMed
-
- Aithal GP, Ramsay L, Daly AK, Sonchit N, Leathart JB, Alexander G, Kenna JG, Caldwell J, Day CP. (2004) Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology .39: 1430–1440 - PubMed
-
- Amar PJ, Schiff ER. (2007) Acetaminophen safety and hepatotoxicity—where do we go from here? Expert Opin Drug Saf .6: 341–355 - PubMed
-
- Andrade RJ, Lucena MI, Fernández MC, Pelaez G, Pachkoria K, García-Ruiz E, García-Muñoz B, González-Grande R, Pizarro A, Durán JA, et al. (2005) Drug-induced liver injury: an analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology .129: 512–521 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
