

Activin A and follistatin-like 3 determine the susceptibility of heart to ischemic injury
- PMID: 19805648
- PMCID: PMC2764796
- DOI: 10.1161/CIRCULATIONAHA.109.872200
Activin A and follistatin-like 3 determine the susceptibility of heart to ischemic injury
Abstract
Background: Transforming growth factor-beta family cytokines have diverse actions in the maintenance of cardiac homeostasis. Activin A is a member of this family whose regulation and function in heart are not well understood at a molecular level. Follistatin-like 3 (Fstl3) is an extracellular regulator of activin A protein, and its function in the heart is also unknown.
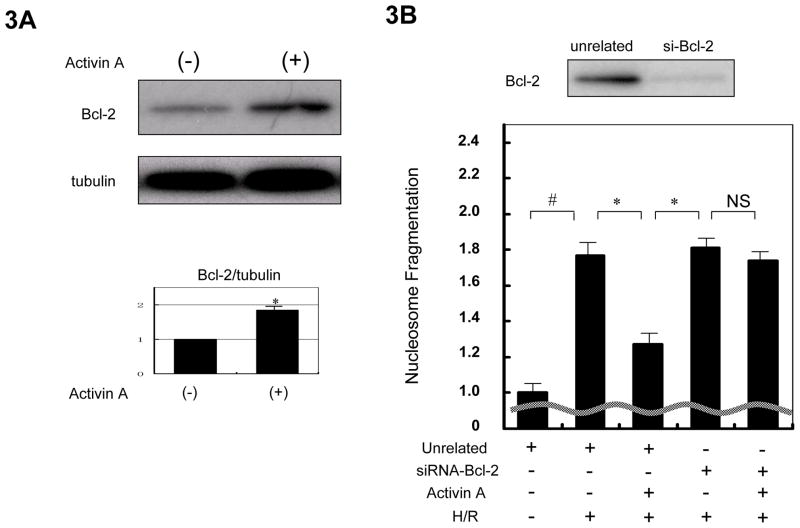
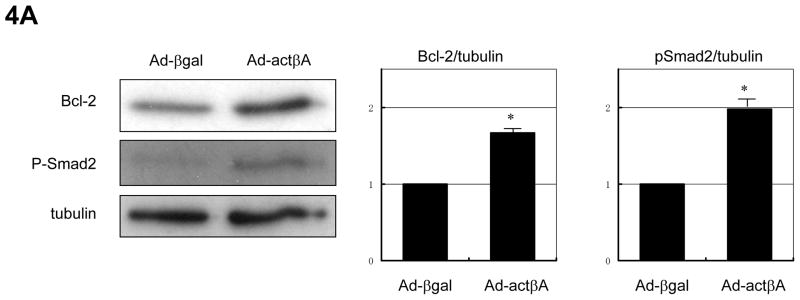
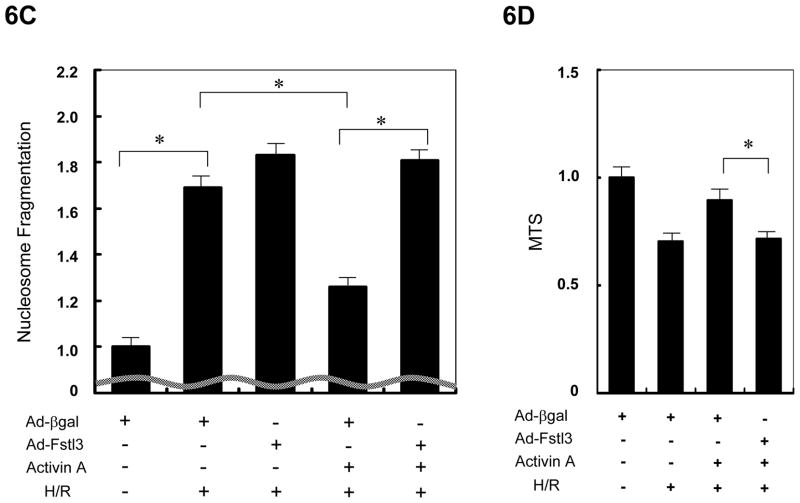
Methods and results: We analyzed the expression of various transforming growth factor-beta superfamily cytokines and their binding partners in mouse heart. Activin betaA and Fstl3 were upregulated in models of myocardial injury. Overexpression of activin A with an adenoviral vector (Ad-actbetaA) or treatment with recombinant activin A protein protected cultured myocytes from hypoxia/reoxygenation-induced apoptosis. Systemic overexpression of activin A in mice by intravenous injection of Ad-actbetaA protected hearts from ischemia/reperfusion injury. Activin A induced the expression of Bcl-2, and ablation of Bcl-2 by small interfering RNA abrogated its protective action in myocytes. The protective effect of activin A on cultured myocytes was abolished by treatment with Fstl3 or by a pharmacological activin receptor-like kinase inhibitor. Cardiac-specific Fstl3 knockout mice showed significantly smaller infarcts after ischemia/reperfusion injury that was accompanied by reduced apoptosis.
Conclusions: Activin A and Fstl3 are induced in heart by myocardial stress. Activin A protects myocytes from death, and this activity is antagonized by Fstl3. Thus, the relative expression levels of these factors after injury is a determinant of cell survival in the heart.
Figures









References
-
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. - PubMed
-
- Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M, Yuge K, Miyata S, Maruyama R, Mikami A, Minatoguchi S, Fujiwara T, Fujiwara H. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–7. - PubMed
-
- Izumi M, Fujio Y, Kunisada K, Negoro S, Tone E, Funamoto M, Osugi T, Oshima Y, Nakaoka Y, Kishimoto T, Yamauchi-Takihara K, Hirota H. Bone morphogenetic protein-2 inhibits serum deprivation-induced apoptosis of neonatal cardiac myocytes through activation of the Smad1 pathway. J Biol Chem. 2001;276:31133–41. - PubMed
-
- Masaki M, Izumi M, Oshima Y, Nakaoka Y, Kuroda T, Kimura R, Sugiyama S, Terai K, Kitakaze M, Yamauchi-Takihara K, Kawase I, Hirota H. Smad1 protects cardiomyocytes from ischemia-reperfusion injury. Circulation. 2005;111:2752–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL77774/HL/NHLBI NIH HHS/United States
- R01 HL086785/HL/NHLBI NIH HHS/United States
- K08 HL071563/HL/NHLBI NIH HHS/United States
- HL71563/HL/NHLBI NIH HHS/United States
- PG/08/084/25827/BHF_/British Heart Foundation/United Kingdom
- R01 AG015052/AG/NIA NIH HHS/United States
- AG15052/AG/NIA NIH HHS/United States
- R37 AG015052/AG/NIA NIH HHS/United States
- P01 HL081587/HL/NHLBI NIH HHS/United States
- U54 AR052646/AR/NIAMS NIH HHS/United States
- HL81587/HL/NHLBI NIH HHS/United States
- R01 HL077774/HL/NHLBI NIH HHS/United States
- HL86785/HL/NHLBI NIH HHS/United States
- U54-AR052646/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
