

Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis
- PMID: 19805911
- PMCID: PMC2769194
- DOI: 10.1172/JCI38674
Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis
Erratum in
- J Clin Invest. 2013 Apr 1;123(4)1844
Abstract
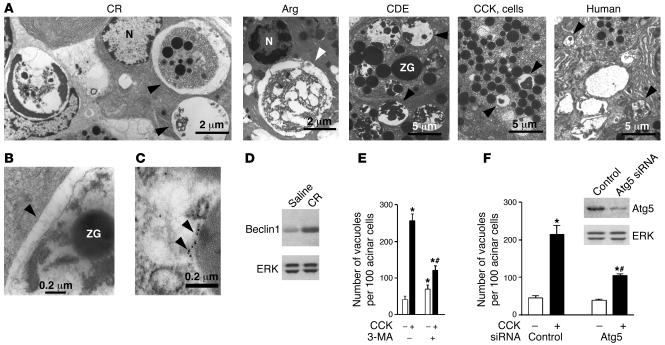
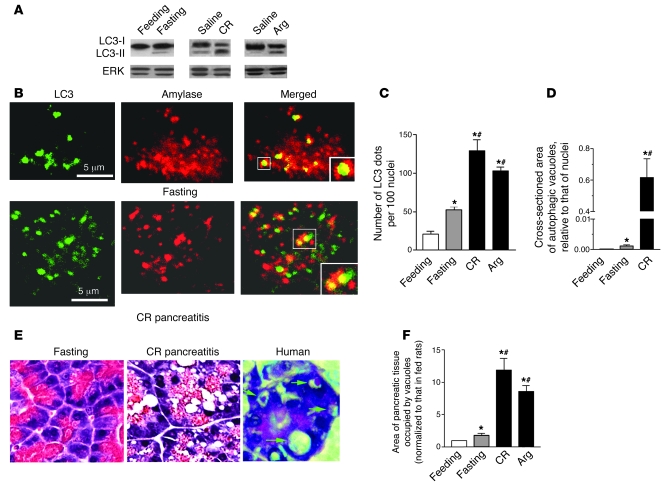
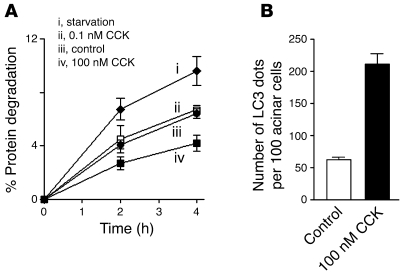
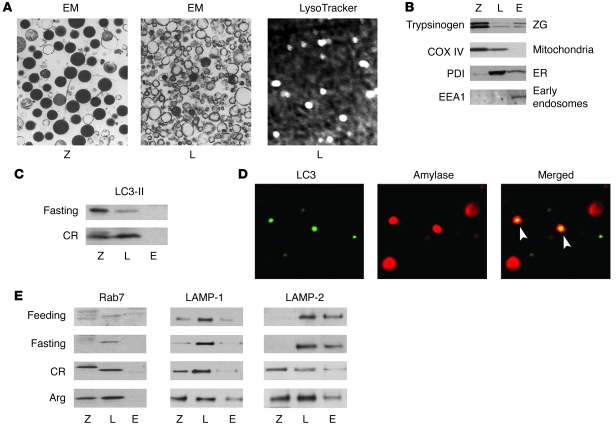
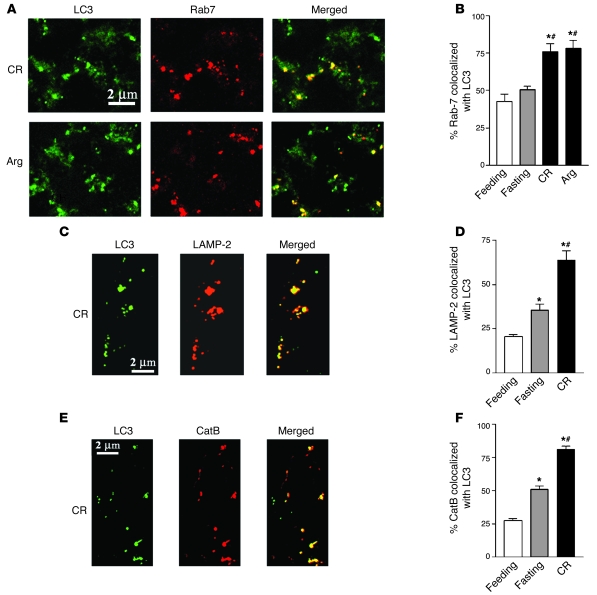
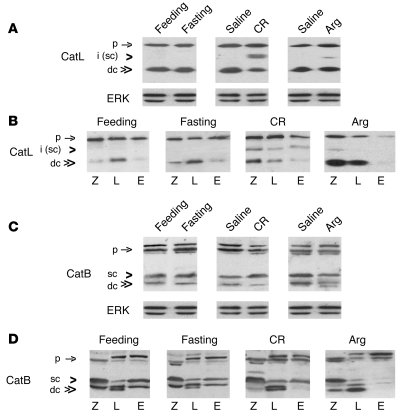
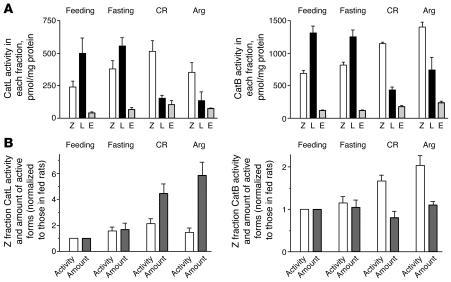
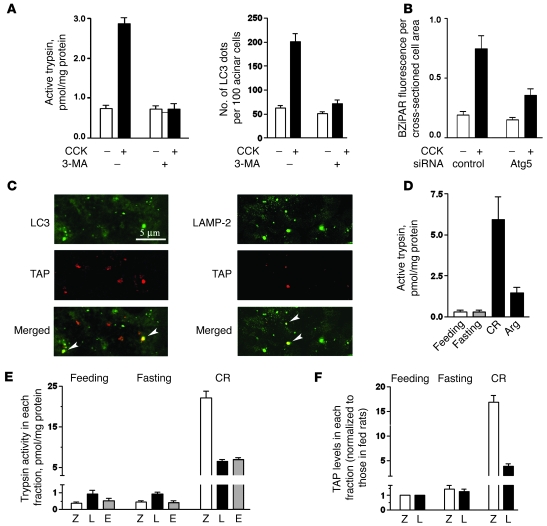
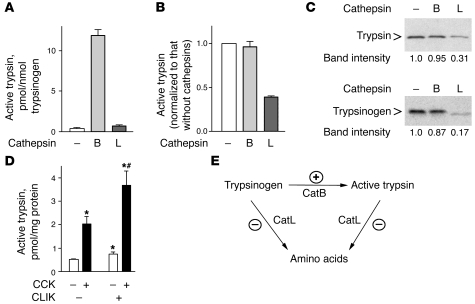
The pathogenic mechanisms underlying acute pancreatitis are not clear. Two key pathologic acinar cell responses of this disease are vacuole accumulation and trypsinogen activation. We show here that both result from defective autophagy, by comparing the autophagic responses in rodent models of acute pancreatitis to physiologic autophagy triggered by fasting. Pancreatitis-induced vacuoles in acinar cells were greater in number and much larger than those induced with fasting. Degradation of long-lived proteins, a measure of autophagic efficiency, was markedly inhibited in in vitro pancreatitis, while it was stimulated by acinar cell starvation. Further, processing of the lysosomal proteases cathepsin L (CatL) and CatB into their fully active, mature forms was reduced in pancreatitis, as were their activities in the lysosome-enriched subcellular fraction. These findings indicate that autophagy is retarded in pancreatitis due to deficient lysosomal degradation caused by impaired cathepsin processing. Trypsinogen activation occurred in pancreatitis but not with fasting and was prevented by inhibiting autophagy. A marker of trypsinogen activation partially localized to autophagic vacuoles, and pharmacologic inhibition of CatL increased the amount of active trypsin in acinar cells. The results suggest that retarded autophagy is associated with an imbalance between CatL, which degrades trypsinogen and trypsin, and CatB, which converts trypsinogen into trypsin, resulting in intra-acinar accumulation of active trypsin in pancreatitis. Thus, deficient lysosomal degradation may be a dominant mechanism for increased intra-acinar trypsin in pancreatitis.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
