

Theory of proton-coupled electron transfer in energy conversion processes
- PMID: 19807148
- PMCID: PMC2841513
- DOI: 10.1021/ar9001284
Theory of proton-coupled electron transfer in energy conversion processes
Abstract
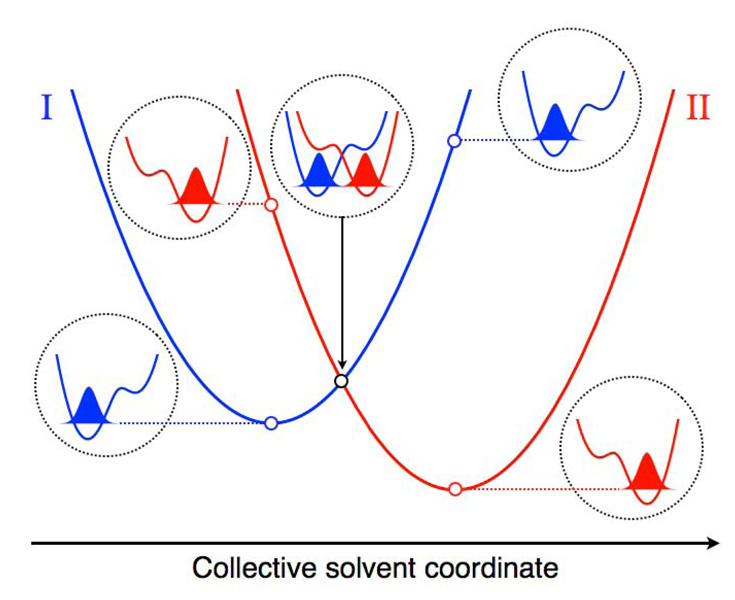
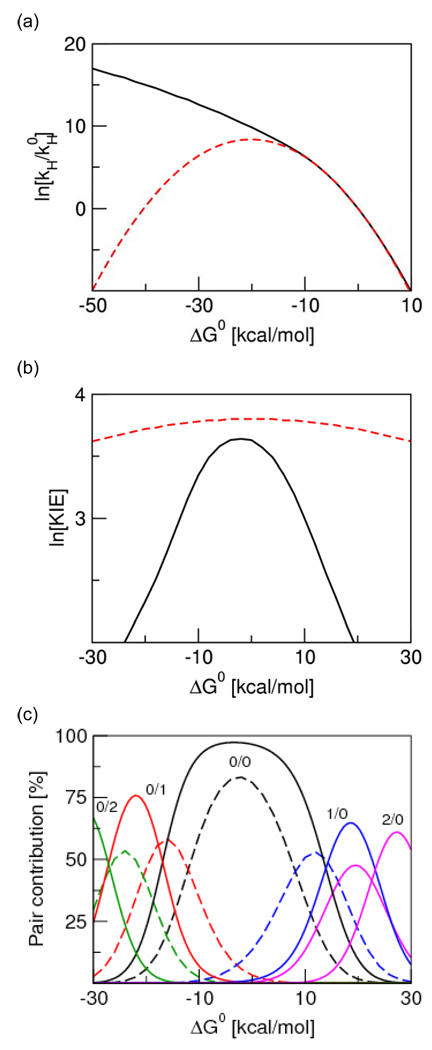
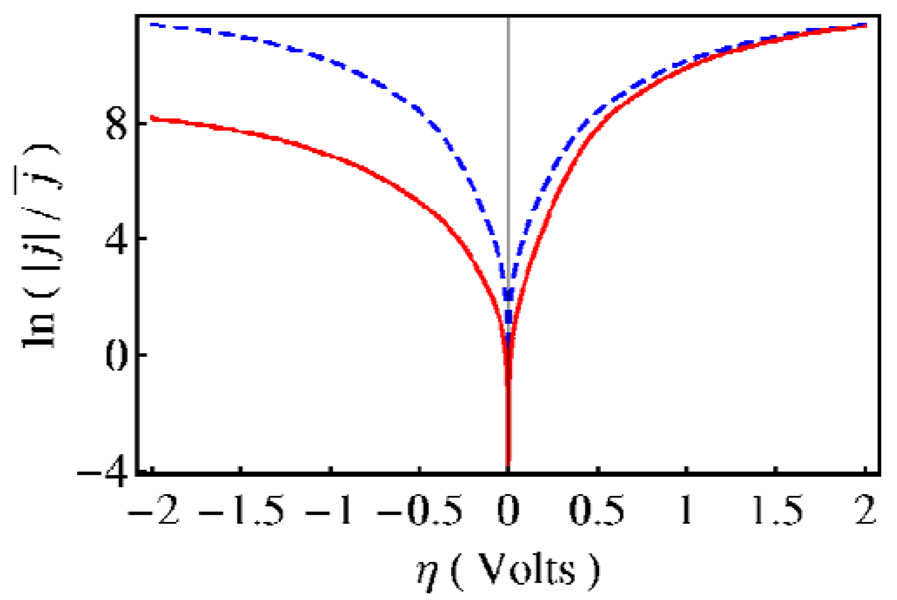
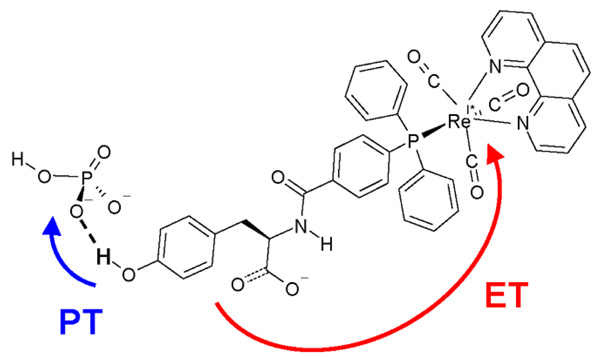
Proton-coupled electron transfer (PCET) reactions play an essential role in a broad range of energy conversion processes, including photosynthesis and respiration. These reactions also form the basis of many types of solar fuel cells and electrochemical devices. Recent advances in the theory of PCET enable the prediction of the impact of system properties on the reaction rates. These predictions may guide the design of more efficient catalysts for energy production, including those based on artificial photosynthesis and solar energy conversion. This Account summarizes the theoretically predicted dependence of PCET rates on system properties and illustrates potential approaches for tuning the reaction rates in chemical systems. A general theoretical formulation for PCET reactions has been developed over the past decade. In this theory, PCET reactions are described in terms of nonadiabatic transitions between the reactant and product electron-proton vibronic states. A series of nonadiabatic rate constant expressions for both homogeneous and electrochemical PCET reactions have been derived in various well-defined limits. Recently this theory has been extended to include the effects of solvent dynamics and to describe ultrafast interfacial PCET. Analysis of the rate constant expressions provides insight into the underlying physical principles of PCET and enables the prediction of the dependence of the rates on the physical properties of the system. Moreover, the kinetic isotope effect, which is the ratio of the rates for hydrogen and deuterium, provides a useful mechanistic probe. Typically the PCET rate will increase as the electronic coupling and temperature increase and as the total reorganization energy and equilibrium proton donor-acceptor distance decrease. The rate constant is predicted to increase as the driving force becomes more negative, rather than exhibit turnover behavior in the inverted region, because excited vibronic product states associated with low free energy barriers and relatively large vibronic couplings become accessible. The physical basis for the experimentally observed pH dependence of PCET reactions has been debated in the literature. When the proton acceptor is a buffer species, the pH dependence may arise from the protonation equilibrium of the buffer. It could also arise from kinetic complexity of competing concerted and sequential PCET reaction pathways. In electrochemical PCET, the heterogeneous rate constants and current densities depend strongly on the overpotential. The change in equilibrium proton donor-acceptor distance upon electron transfer may lead to asymmetries in the Tafel plots and deviations of the transfer coefficient from the standard value of one-half at zero overpotential. Applications of this theory to experimentally studied systems illustrate approaches that can be utilized to tune the PCET rate. For example, the rate can be tuned by changing the pH or using different buffer species as proton acceptors. The rate can also be tuned with site-specific mutagenesis in biological systems or chemical modifications that vary the substituents on the redox species in chemical systems. Understanding the impact of these changes on the PCET rate may assist experimental efforts to enhance energy conversion processes.
Figures
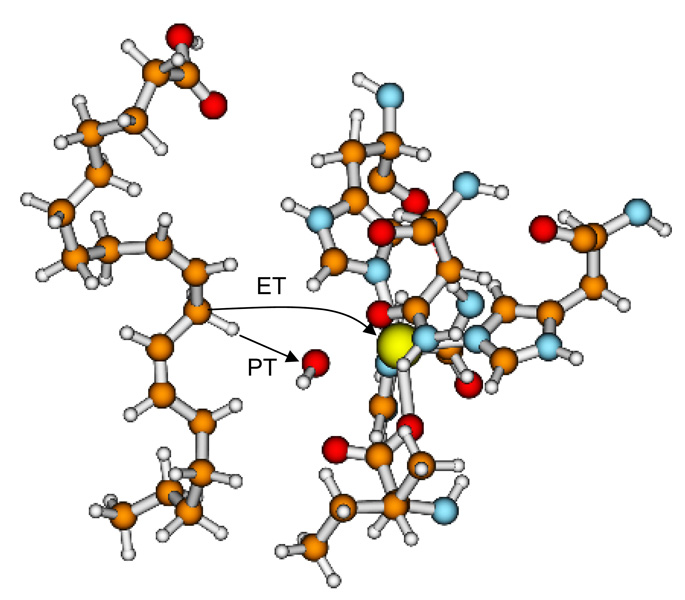
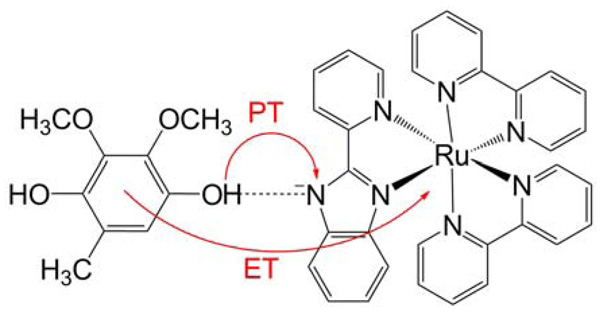
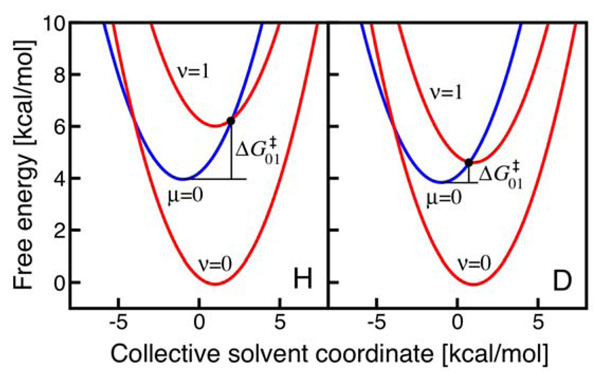
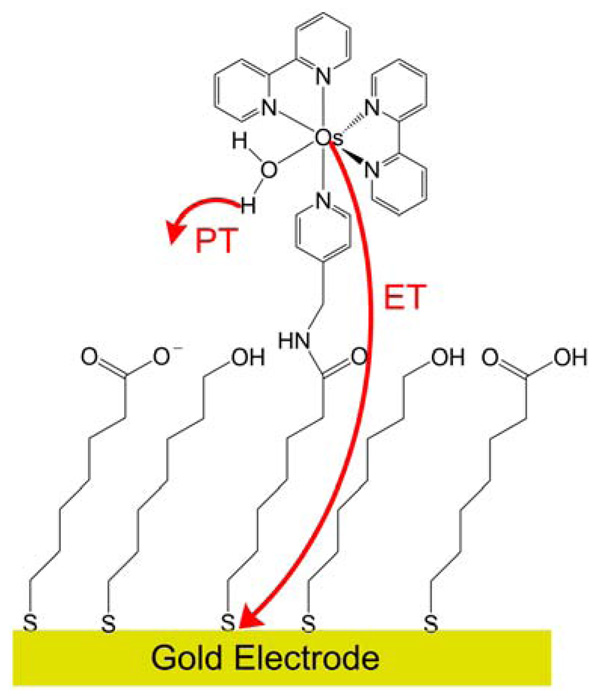
Similar articles
-
Explaining Kinetic Isotope Effects in Proton-Coupled Electron Transfer Reactions.Acc Chem Res. 2025 Apr 15;58(8):1335-1344. doi: 10.1021/acs.accounts.5c00119. Epub 2025 Apr 4. Acc Chem Res. 2025. PMID: 40184268
-
Analysis of kinetic isotope effects for proton-coupled electron transfer reactions.J Phys Chem A. 2009 Mar 12;113(10):2117-26. doi: 10.1021/jp809122y. J Phys Chem A. 2009. PMID: 19182970 Free PMC article.
-
Buffer-assisted proton-coupled electron transfer in a model rhenium-tyrosine complex.J Am Chem Soc. 2007 Sep 12;129(36):11146-52. doi: 10.1021/ja072708k. Epub 2007 Aug 18. J Am Chem Soc. 2007. PMID: 17705482
-
Proton-coupled electron transfer in solution, proteins, and electrochemistry.J Phys Chem B. 2008 Nov 13;112(45):14108-23. doi: 10.1021/jp805876e. Epub 2008 Oct 9. J Phys Chem B. 2008. PMID: 18842015 Free PMC article. Review.
-
Theoretical Modeling of Electrochemical Proton-Coupled Electron Transfer.Chem Rev. 2022 Jun 22;122(12):10599-10650. doi: 10.1021/acs.chemrev.1c00929. Epub 2022 Mar 1. Chem Rev. 2022. PMID: 35230812 Review.
Cited by
-
Photochemical Tyrosine Oxidation with a Hydrogen-Bonded Proton Acceptor by Bidirectional Proton-Coupled Electron Transfer.Chem Sci. 2012 Aug;3(8):2457-2461. doi: 10.1039/C2SC20113E. Chem Sci. 2012. PMID: 23495362 Free PMC article.
-
In Silico Studies of Small Molecule Interactions with Enzymes Reveal Aspects of Catalytic Function.Catalysts. 2017;7(7):212. doi: 10.3390/catal7070212. Epub 2017 Jul 14. Catalysts. 2017. PMID: 30464857 Free PMC article.
-
Solvent-modulated proton-coupled electron transfer in an iridium complex with an ESIPT ligand.Chem Sci. 2022 Mar 5;13(13):3809-3818. doi: 10.1039/d1sc07250a. eCollection 2022 Mar 30. Chem Sci. 2022. PMID: 35432886 Free PMC article.
-
Multifaceted aspects of charge transfer.Phys Chem Chem Phys. 2020 Oct 14;22(38):21583-21629. doi: 10.1039/d0cp01556c. Epub 2020 Aug 12. Phys Chem Chem Phys. 2020. PMID: 32785306 Free PMC article.
-
Efficient tri-metallic oxides NiCo2O4/CuO for the oxygen evolution reaction.RSC Adv. 2019 Dec 20;9(72):42387-42394. doi: 10.1039/c9ra09351f. eCollection 2019 Dec 18. RSC Adv. 2019. PMID: 35542865 Free PMC article.
References
-
- Cukier RI, Nocera DG. Proton-coupled electron transfer. Annu. Rev. Phys. Chem. 1998;49:337–369. - PubMed
-
- Hammes-Schiffer S. Theoretical perspectives on proton-coupled electron transfer reactions. Acc. Chem. Res. 2001;34:273–281. - PubMed
-
- Mayer JM. Proton-coupled electron transfer: A reaction chemist's view. Annu. Rev. Phys. Chem. 2004;55:363–390. - PubMed
-
- Rosenthal J, Nocera DG. Role of proton-coupled electron transfer in O-O bond activation. Acc. Chem. Res. 2007;40:543–553. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
