

Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data
- PMID: 19808877
- PMCID: PMC2788925
- DOI: 10.1093/bioinformatics/btp579
Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data
Abstract
Motivation: Next-generation sequencing has become an important tool for genome-wide quantification of DNA and RNA. However, a major technical hurdle lies in the need to map short sequence reads back to their correct locations in a reference genome. Here, we investigate the impact of SNP variation on the reliability of read-mapping in the context of detecting allele-specific expression (ASE).
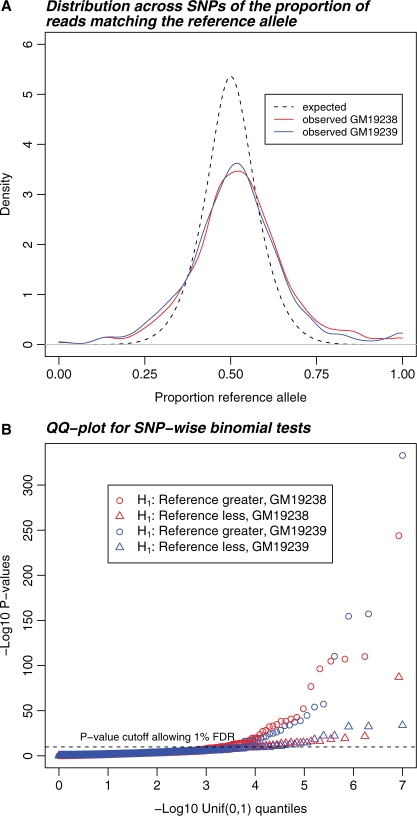
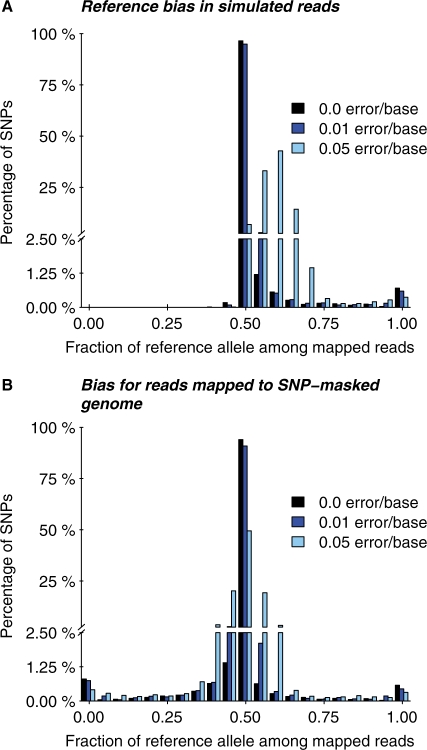
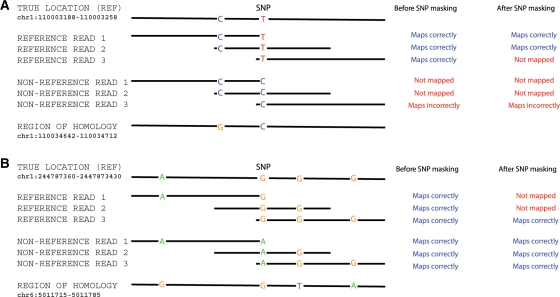
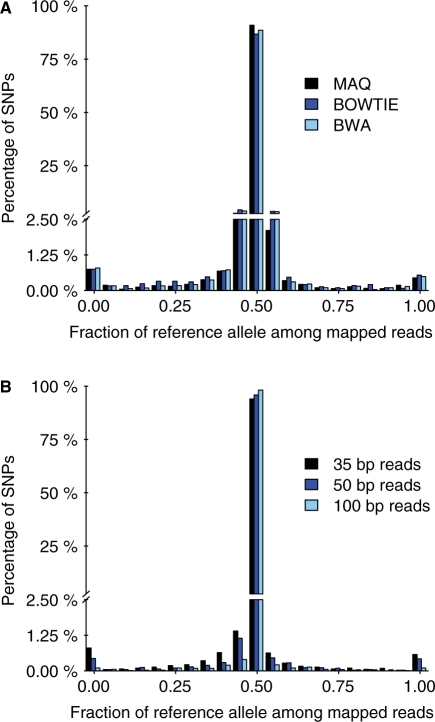
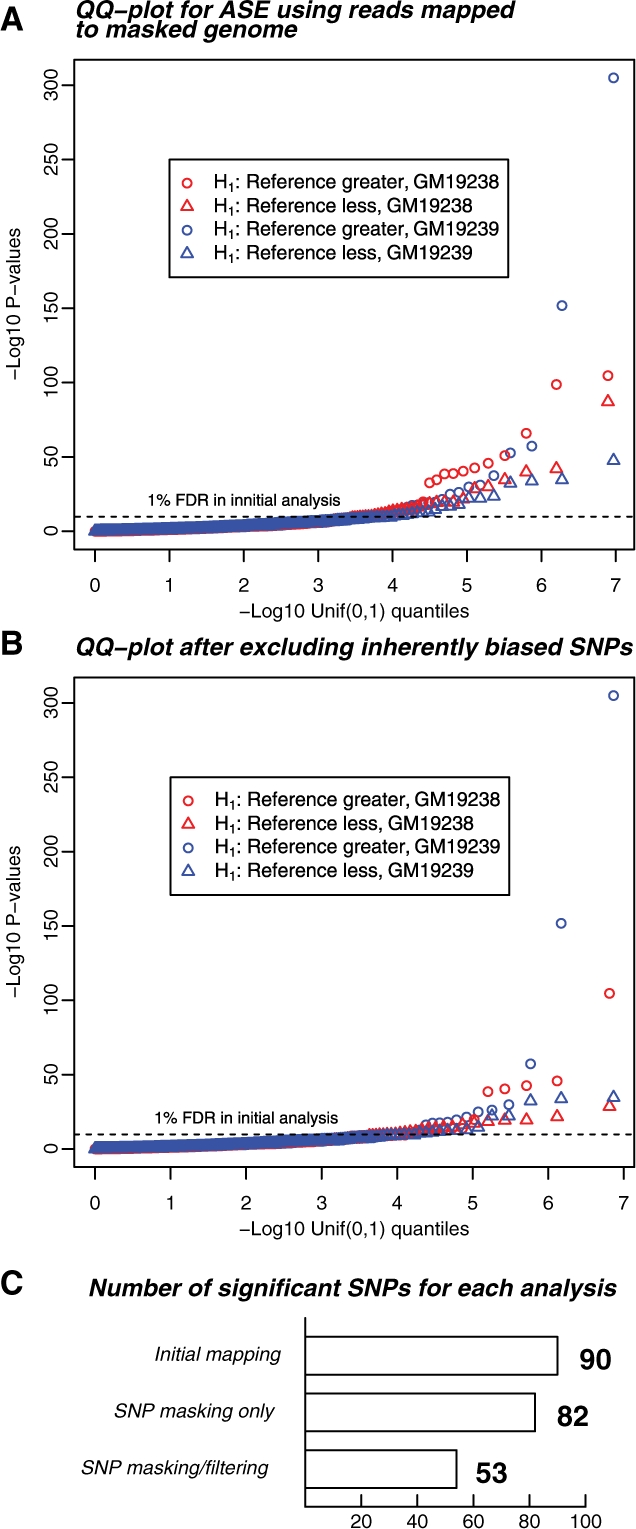
Results: We generated 16 million 35 bp reads from mRNA of each of two HapMap Yoruba individuals. When we mapped these reads to the human genome we found that, at heterozygous SNPs, there was a significant bias toward higher mapping rates of the allele in the reference sequence, compared with the alternative allele. Masking known SNP positions in the genome sequence eliminated the reference bias but, surprisingly, did not lead to more reliable results overall. We find that even after masking, approximately 5-10% of SNPs still have an inherent bias toward more effective mapping of one allele. Filtering out inherently biased SNPs removes 40% of the top signals of ASE. The remaining SNPs showing ASE are enriched in genes previously known to harbor cis-regulatory variation or known to show uniparental imprinting. Our results have implications for a variety of applications involving detection of alternate alleles from short-read sequence data.
Availability: Scripts, written in Perl and R, for simulating short reads, masking SNP variation in a reference genome and analyzing the simulation output are available upon request from JFD. Raw short read data were deposited in GEO (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE18156.
Contact: jdegner@uchicago.edu; marioni@uchicago.edu; gilad@uchicago.edu; pritch@uchicago.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
-
- Babak T, et al. Global survey of genomic imprinting by transcriptome sequencing. Curr. Biol. 2008;18:1735–1741. - PubMed
-
- Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am. J. Med. Genet. A. 2008;146A:2041–2052. - PubMed
-
- Kaiser J. DNA sequencing. A plan to capture human diversity in 1000 genomes. Science. 2008;319:395. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
