

DNA cleavage and methylation specificity of the single polypeptide restriction-modification enzyme LlaGI
- PMID: 19808936
- PMCID: PMC2790903
- DOI: 10.1093/nar/gkp790
DNA cleavage and methylation specificity of the single polypeptide restriction-modification enzyme LlaGI
Abstract
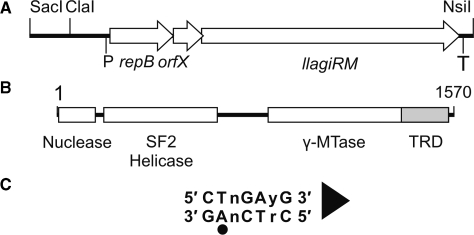
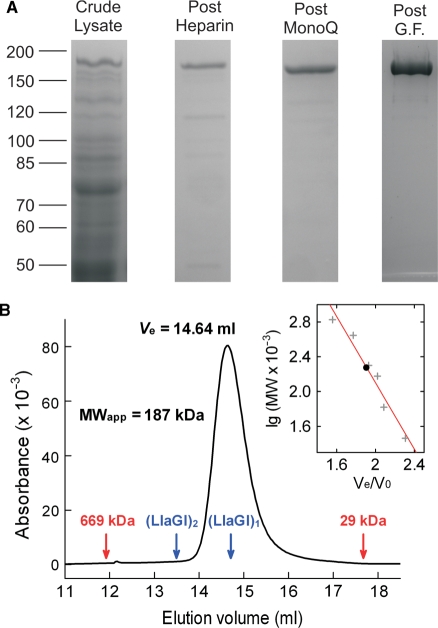
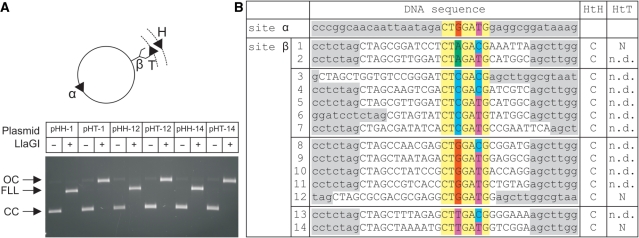
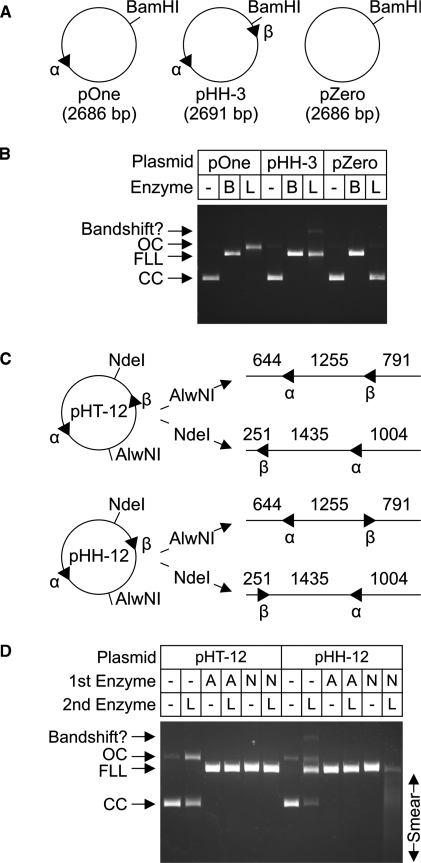
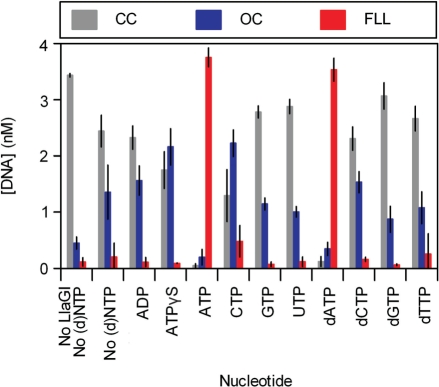
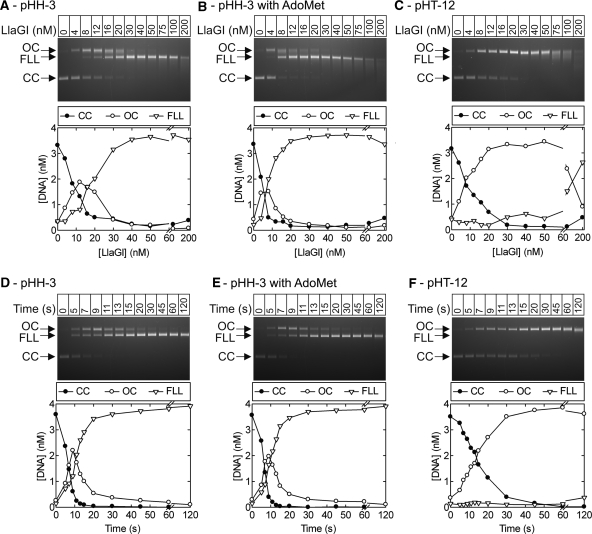
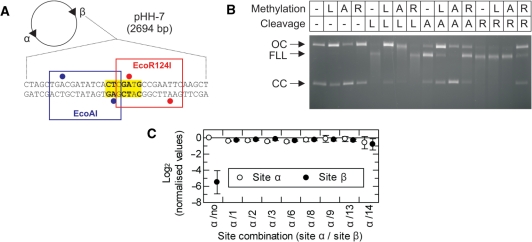
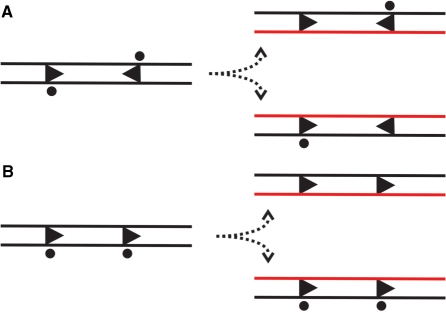
LlaGI is a single polypeptide restriction-modification enzyme encoded on the naturally-occurring plasmid pEW104 isolated from Lactococcus lactis ssp. cremoris W10. Bioinformatics analysis suggests that the enzyme contains domains characteristic of an mrr endonuclease, a superfamily 2 DNA helicase and a gamma-family adenine methyltransferase. LlaGI was expressed and purified from a recombinant clone and its properties characterised. An asymmetric recognition sequence was identified, 5'-CTnGAyG-3' (where n is A, G, C or T and y is C or T). Methylation of the recognition site occurred on only one strand (the non-degenerate dA residue of 5'-CrTCnAG-3' being methylated at the N6 position). Double strand DNA breaks at distant, random sites were only observed when two head-to-head oriented, unmethylated copies of the site were present; single sites or pairs in tail-to-tail or head-to-tail repeat only supported a DNA nicking activity. dsDNA nuclease activity was dependent upon the presence of ATP or dATP. Our results are consistent with a directional long-range communication mechanism that is necessitated by the partial site methylation. In the accompanying manuscript [Smith et al. (2009) The single polypeptide restriction-modification enzyme LlaGI is a self-contained molecular motor that translocates DNA loops], we demonstrate that this communication is via 1-dimensional DNA loop translocation. On the basis of this data and that in the third accompanying manuscript [Smith et al. (2009) An Mrr-family nuclease motif in the single polypeptide restriction-modification enzyme LlaGI], we propose that LlaGI is the prototype of a new sub-classification of Restriction-Modification enzymes, named Type I SP (for Single Polypeptide).
Figures
References
-
- Madsen A, Josephsen J. The LlaGI restriction and modification system of Lactococcus lactis W10 consists of only one single polypeptide. FEMS Microbiol. Lett. 2001;200:91–96. - PubMed
-
- Bujnicki JM, Rychlewski L. Identification of a PD-(D/E)XK-like domain with a novel configuration of the endonuclease active site in the methyl-directed restriction enzyme Mrr and its homologs. Gene. 2001;267:183–191. - PubMed
-
- Bourniquel AA, Bickle TA. Complex restriction enzymes: NTP-driven molecular motors. Biochimie. 2002;84:1047–1059. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
