

An evaluation of statistical approaches to rare variant analysis in genetic association studies
- PMID: 19810025
- PMCID: PMC2962811
- DOI: 10.1002/gepi.20450
An evaluation of statistical approaches to rare variant analysis in genetic association studies
Abstract
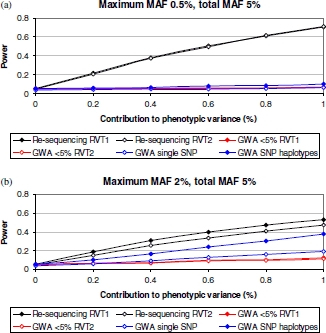
Genome-wide association (GWA) studies have proved to be extremely successful in identifying novel common polymorphisms contributing effects to the genetic component underlying complex traits. Nevertheless, one source of, as yet, undiscovered genetic determinants of complex traits are those mediated through the effects of rare variants. With the increasing availability of large-scale re-sequencing data for rare variant discovery, we have developed a novel statistical method for the detection of complex trait associations with these loci, based on searching for accumulations of minor alleles within the same functional unit. We have undertaken simulations to evaluate strategies for the identification of rare variant associations in population-based genetic studies when data are available from re-sequencing discovery efforts or from commercially available GWA chips. Our results demonstrate that methods based on accumulations of rare variants discovered through re-sequencing offer substantially greater power than conventional analysis of GWA data, and thus provide an exciting opportunity for future discovery of genetic determinants of complex traits.
2009 Wiley-Liss, Inc.
Figures
References
-
- Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, Penninx BW, Janssens AC, Wilson JF, Spector T, Martin NG, Pedersen NL, Kyvik KO, Kaprio T, Hofman A, Freimer NB, Jarvelin MR, Gyllensten U, Campbell H, Rudan I, Johansson A, Marroni F, Hayward C, Vitart V, Jonasson I, Pattaro C, Wright A, Hastle N, Pichler I, Hicks AA, Falchi M, Willemsen G, Hottenga JJ, de Geus EJ, Montgomery GW, Whitfield J, Magnusson P, Sabarinen J, Perola M, Silander K, Isaacs A, Sijbrands EJ, Uitterlindcn AG, Witteman JC, Oostra BA, Elliott P, Ruokonen A, Sabatti C, Gieger C, Meitinger T, Kronenberg F, Doring A, Wichmann HE, Smit JH, McCarthy MI, van Duijn CM, Peltonen L. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41:47–55. - PMC - PubMed
-
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, NIDDK IBD Genetics Consortium. Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot TP, de Vos M, Vermeire S, Louis E, Belgian-French IBD Consortium; Wellcome Trust Case Control Consortium. Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori T, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MT. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–962. - PMC - PubMed
-
- Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
