

Caring for Machado-Joseph disease: current understanding and how to help patients
- PMID: 19811945
- PMCID: PMC2818316
- DOI: 10.1016/j.parkreldis.2009.08.012
Caring for Machado-Joseph disease: current understanding and how to help patients
Abstract
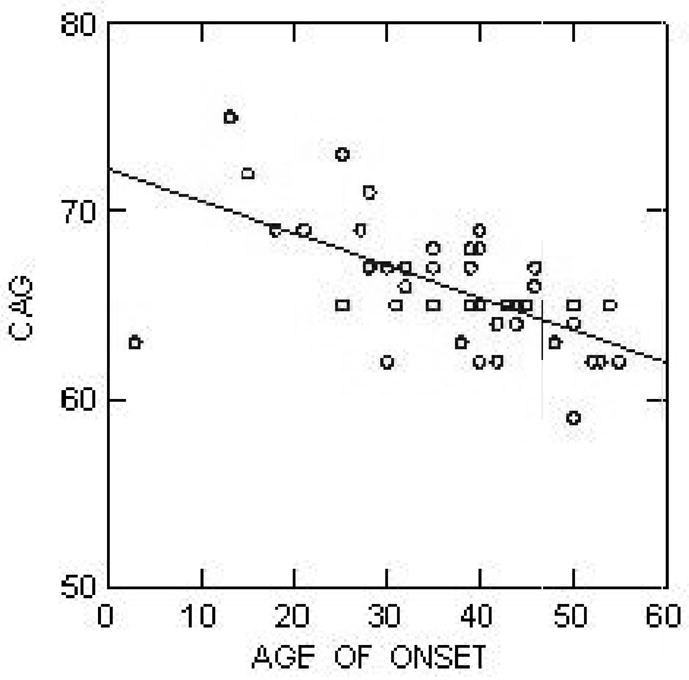
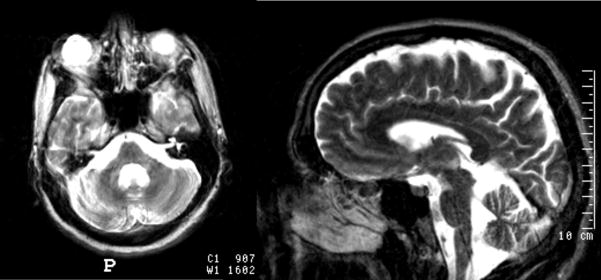
Machado-Joseph disease or spinocerebellar ataxia 3 (MJD/SCA3) is a clinically heterogeneous, neurodegenerative disorder characterized by varying degrees of ataxia, ophthalmoplegia, peripheral neuropathy, pyramidal dysfunction and movement disorder. MJD/SCA3 is caused by a CAG repeat expansion mutation in the protein coding region of the ATXN3 gene located at chromosome 14q32.1. Current hypotheses regarding pathogenesis favor the view that mutated ataxin-3, with its polyglutamine expansion, is prone to adopt an abnormal conformation, engage in altered protein-protein interactions and aggregate. Expanded CAG repeat length correlates with the range and severity of the clinical manifestations and inversely correlates with age of disease onset. Though MJD/SCA3 is classically described as affecting the cerebellum, brainstem and basal ganglia, recent neuropathology and neuroimaging series demonstrate involvement of other areas such as the thalamus and cerebral cortex. Clinically, much emphasis has been placed in the description and recognition of the non-motor symptoms observed in these patients, such as pain, cramps, fatigue and depression. Currently, no disease modifying treatment exists for MJD/SCA3. Standard of care includes genetic counseling, exercise/physical therapy programs, and speech and swallow evaluation. Symptomatic treatment for clinical findings such as depression, sleep disorders, parkinsonism, dystonia, cramps, and pain is important to improve the quality of life for those with MJD/SCA3.
Figures
References
-
- Nakano KK, Dawson DM, Spence A. Machado disease. A hereditary ataxia in Portuguese emigrants to Massachusetts. Neurology. 1972;22:49–55. - PubMed
-
- Woods BT, Schaumburg HH. Nigro-spino-dentatal degeneration with nuclear ophthalmoplegia: a unique and partially treatable clinico-pathological entity. J Neurol Sci. 1972;17:149–166. - PubMed
-
- Rosenberg RN, Nyhan WL, Bay C, Shore P. Autosomal dominant striato-nigral degeneration: a clinical, pathologic and biochemical study of a new genetic disorder. Neurology. 1976;26:703–714. - PubMed
-
- Romanul FCA, Fowler HL, Radvany J, Feldman RG, Feingold M. Azorean disease of the nervous system. New Eng J Med. 1977;296:1505–1508. - PubMed
-
- Rosenberg RN. Machado-Joseph disease: an autosomal dominant motor system degeneration. Mov Disord. 1992;7:193–203. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
