

A simple method for directional transcriptome sequencing using Illumina technology
- PMID: 19815668
- PMCID: PMC2794173
- DOI: 10.1093/nar/gkp811
A simple method for directional transcriptome sequencing using Illumina technology
Abstract
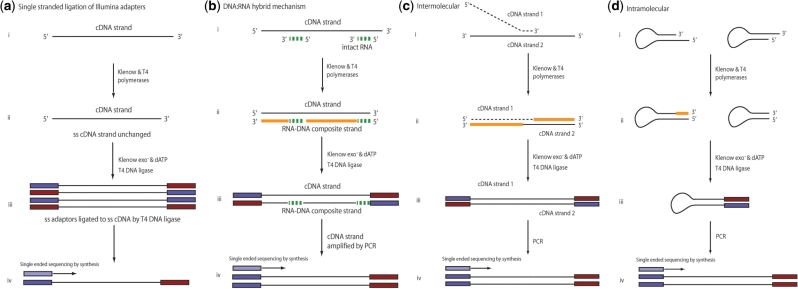
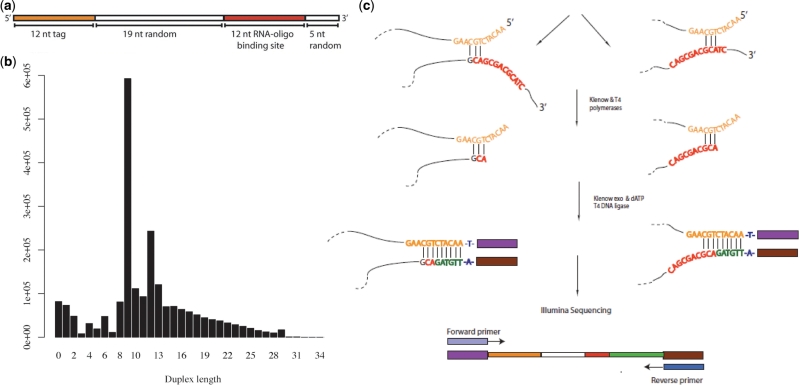
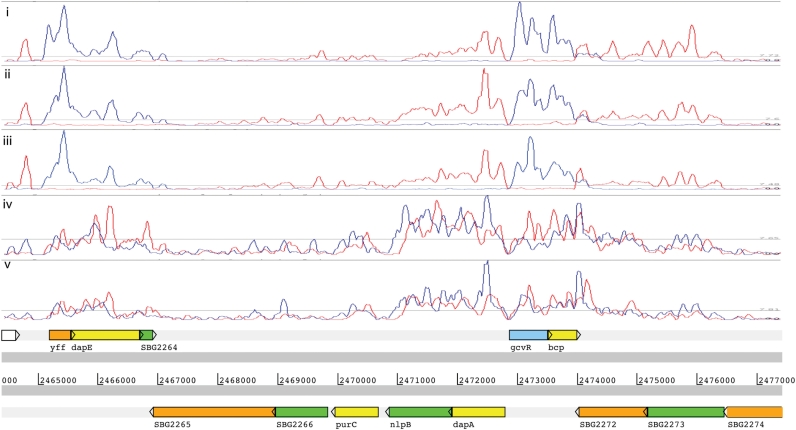
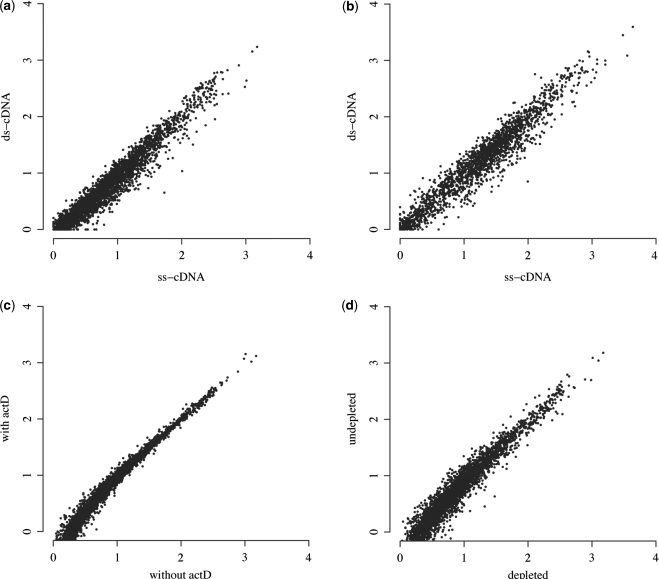
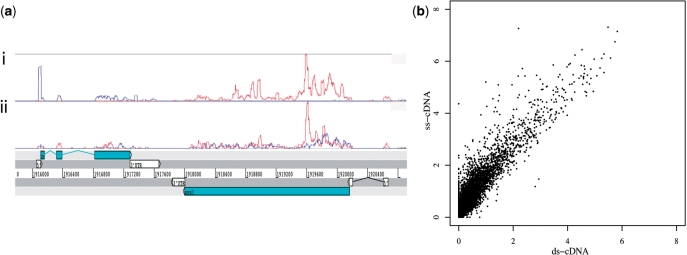
High-throughput sequencing of cDNA has been used to study eukaryotic transcription on a genome-wide scale to single base pair resolution. In order to compensate for the high ribonuclease activity in bacterial cells, we have devised an equivalent technique optimized for studying complete prokaryotic transcriptomes that minimizes the manipulation of the RNA sample. This new approach uses Illumina technology to sequence single-stranded (ss) cDNA, generating information on both the direction and level of transcription throughout the genome. The protocol, and associated data analysis programs, are freely available from http://www.sanger.ac.uk/Projects/Pathogens/Transcriptome/. We have successfully applied this method to the bacterial pathogens Salmonella bongori and Streptococcus pneumoniae and the yeast Schizosaccharomyces pombe. This method enables experimental validation of genetic features predicted in silico and allows the easy identification of novel transcripts throughout the genome. We also show that there is a high correlation between the level of gene expression calculated from ss-cDNA and double-stranded-cDNA sequencing, indicting that ss-cDNA sequencing is both robust and appropriate for use in quantitative studies of transcription. Hence, this simple method should prove a useful tool in aiding genome annotation and gene expression studies in both prokaryotes and eukaryotes.
Figures
References
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. - PubMed
-
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. - PubMed
-
- Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bahler J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
