

Modeling structure-function relationships in synthetic DNA sequences using attribute grammars
- PMID: 19816554
- PMCID: PMC2748682
- DOI: 10.1371/journal.pcbi.1000529
Modeling structure-function relationships in synthetic DNA sequences using attribute grammars
Abstract
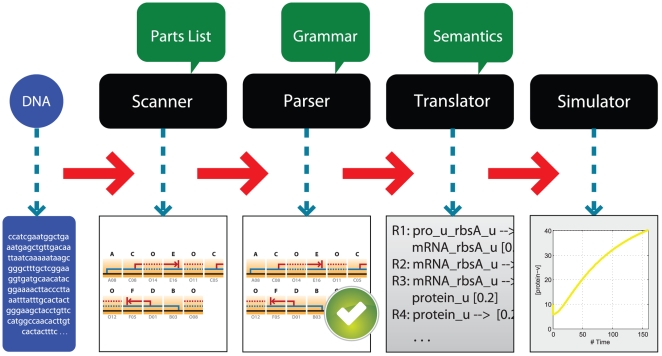
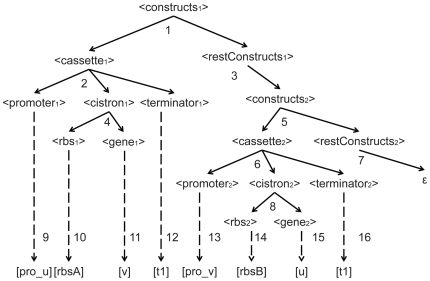
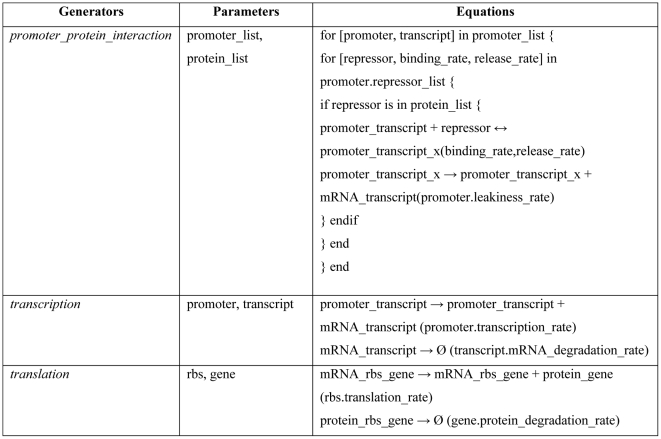
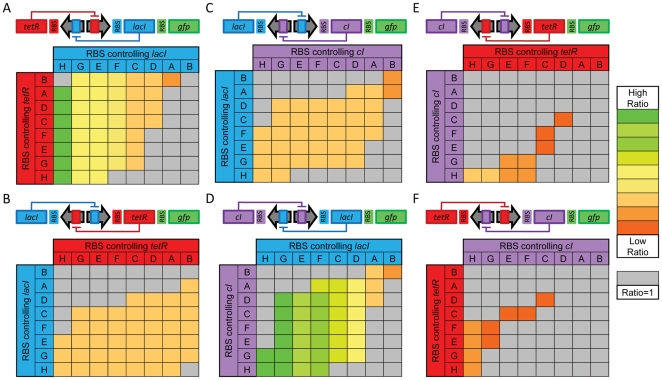
Recognizing that certain biological functions can be associated with specific DNA sequences has led various fields of biology to adopt the notion of the genetic part. This concept provides a finer level of granularity than the traditional notion of the gene. However, a method of formally relating how a set of parts relates to a function has not yet emerged. Synthetic biology both demands such a formalism and provides an ideal setting for testing hypotheses about relationships between DNA sequences and phenotypes beyond the gene-centric methods used in genetics. Attribute grammars are used in computer science to translate the text of a program source code into the computational operations it represents. By associating attributes with parts, modifying the value of these attributes using rules that describe the structure of DNA sequences, and using a multi-pass compilation process, it is possible to translate DNA sequences into molecular interaction network models. These capabilities are illustrated by simple example grammars expressing how gene expression rates are dependent upon single or multiple parts. The translation process is validated by systematically generating, translating, and simulating the phenotype of all the sequences in the design space generated by a small library of genetic parts. Attribute grammars represent a flexible framework connecting parts with models of biological function. They will be instrumental for building mathematical models of libraries of genetic constructs synthesized to characterize the function of genetic parts. This formalism is also expected to provide a solid foundation for the development of computer assisted design applications for synthetic biology.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures


References
-
- Keller EF. The century of the gene. Cambridge, Mass.: Harvard University Press; 2000. 186
-
- Sturtevant AH. A history of genetics. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2001. 174
-
- Singer M, Berg P. Timeline - George Beadle: from genes to proteins. Nat Rev Genet. 2004;5:949–954. - PubMed
-
- Tatum EL. A case history in biological research. Science. 1959;129:1711–1715. - PubMed
-
- Lynch M, Walsh B. Genetics and analysis of quantitative traits. Sunderland: Sinauer; 1998. 980
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
