

Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism
- PMID: 19820032
- PMCID: PMC2775659
- DOI: 10.1210/jc.2009-0179
Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism
Abstract
Context: FGFR1 mutations have been identified in about 10% of patients with Kallmann syndrome. Recently cases of idiopathic hypogonadotropic hypogonadism (IHH) with a normal sense of smell (nIHH) have been reported.
Aims: The objective of the study was to define the frequency of FGFR1 mutations in a large cohort of nIHH, delineate the spectrum of reproductive phenotypes, assess functionality of the FGFR1 mutant alleles in vitro, and investigate genotype-phenotype relationships.
Design: FGFR1 sequencing of 134 well-characterized nIHH patients (112 men and 22 women) and 270 healthy controls was performed. The impact of the identified mutations on FGFR1 function was assessed using structural prediction and in vitro studies.
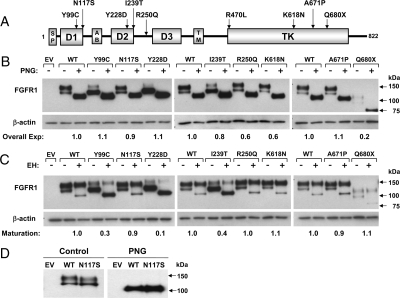
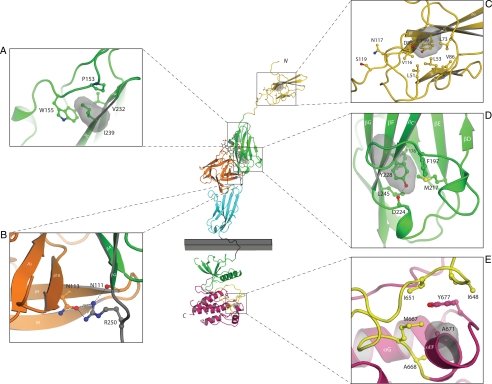
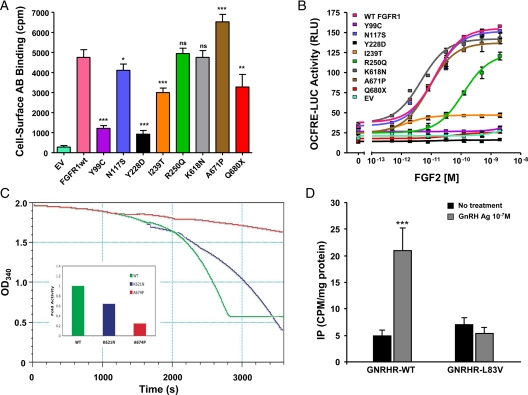
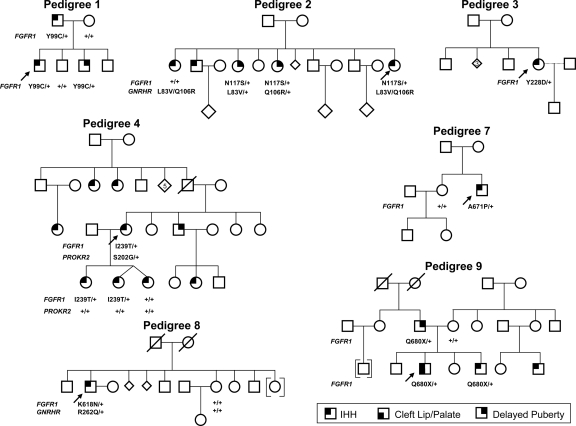
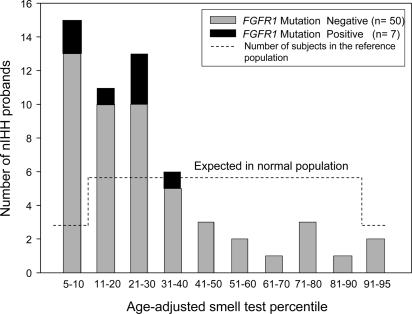
Results: Nine nIHH subjects (five males and four females; 7%) harbor a heterozygous mutation in FGFR1 and exhibit a wide spectrum of pubertal development, ranging from absent puberty to reversal of IHH in both sexes. All mutations impair receptor function. The Y99C, Y228D, and I239T mutants impair the tertiary folding, resulting in incomplete glycosylation and reduced cell surface expression. The R250Q mutant reduces receptor affinity for FGF. The K618N, A671P, and Q680X mutants impair tyrosine kinase activity. However, the degree of functional impairment of the mutant receptors did not always correlate with the reproductive phenotype, and variable expressivity of the disease was noted within family members carrying the same FGFR1 mutation. These discrepancies were partially explained by additional mutations in known IHH loci.
Conclusions: Loss-of-function mutations in FGFR1 underlie 7% of nIHH with different degrees of impairment in vitro. These mutations act in concert with other gene defects in several cases, consistent with oligogenicity.
Figures
References
-
- Santoro N, Filicori M, Crowley Jr WF 1986 Hypogonadotropic disorders in men and women: diagnosis and therapy with pulsatile gonadotropin-releasing hormone. Endocr Rev 7:11–23 - PubMed
-
- Seminara SB, Hayes FJ, Crowley Jr WF 1998 Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann’s syndrome): pathophysiological and genetic considerations. Endocr Rev 19:521–539 - PubMed
-
- Quinton R, Duke VM, Robertson A, Kirk JM, Matfin G, de Zoysa PA, Azcona C, MacColl GS, Jacobs HS, Conway GS, Besser M, Stanhope RG, Bouloux PM 2001 Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf) 55:163–174 - PubMed
-
- Sato N, Katsumata N, Kagami M, Hasegawa T, Hori N, Kawakita S, Minowada S, Shimotsuka A, Shishiba Y, Yokozawa M, Yasuda T, Nagasaki K, Hasegawa D, Hasegawa Y, Tachibana K, Naiki Y, Horikawa R, Tanaka T, Ogata T 2004 Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab 89:1079–1088 - PubMed
-
- Kim SH, Hu Y, Cadman S, Bouloux P 2008 Diversity in fibroblast growth factor receptor 1 regulation: learning from the investigation of Kallmann syndrome. J Neuroendocrinol 20:141–163 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
