

Familial adenomatous polyposis
- PMID: 19822006
- PMCID: PMC2772987
- DOI: 10.1186/1750-1172-4-22
Familial adenomatous polyposis
Abstract
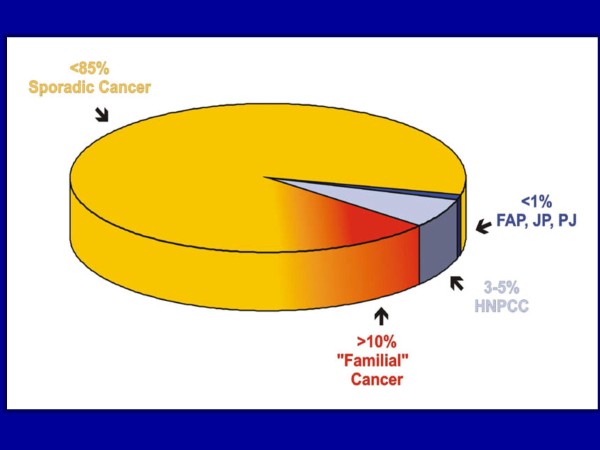
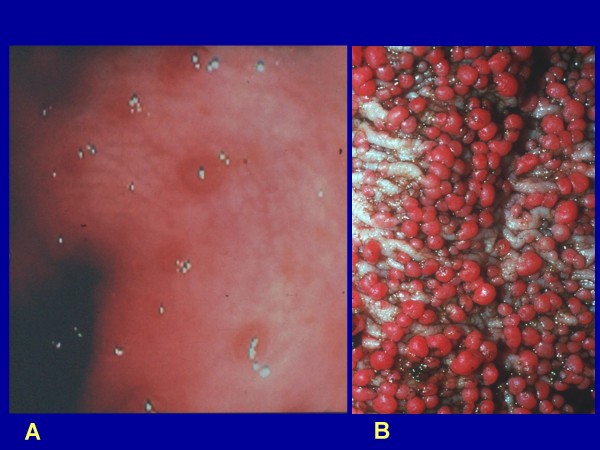
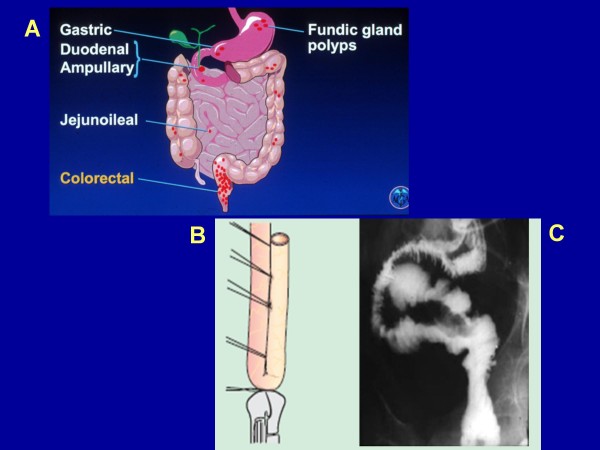
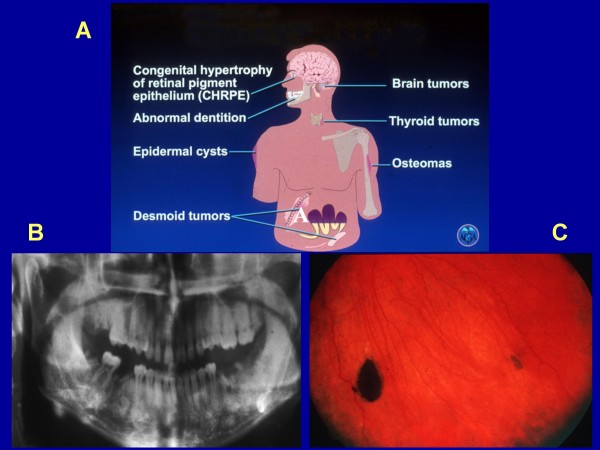
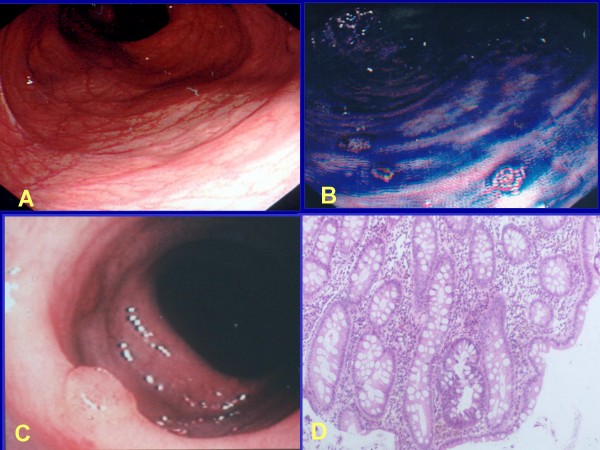
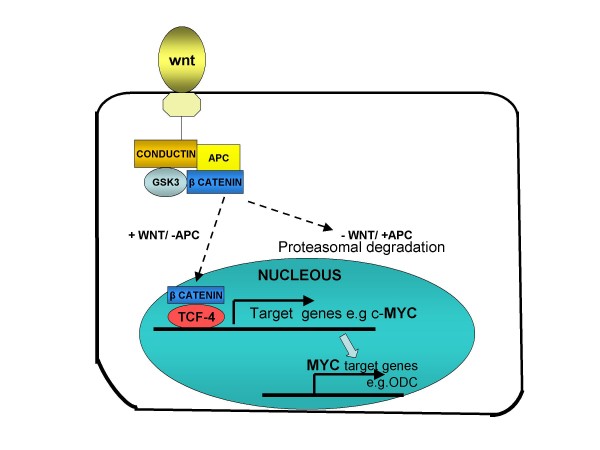
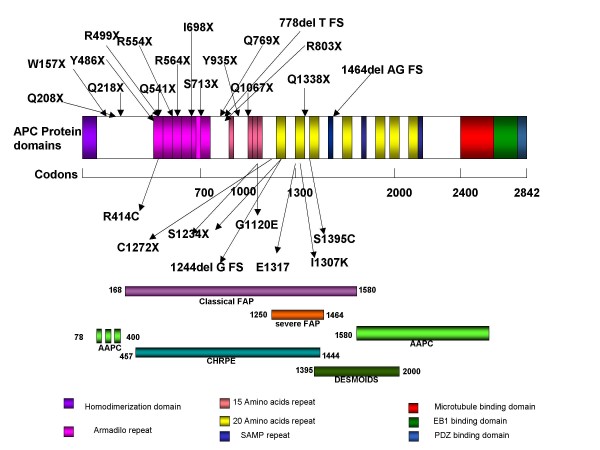
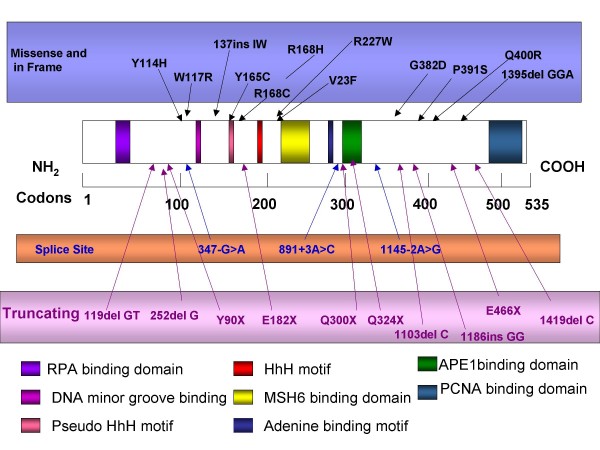
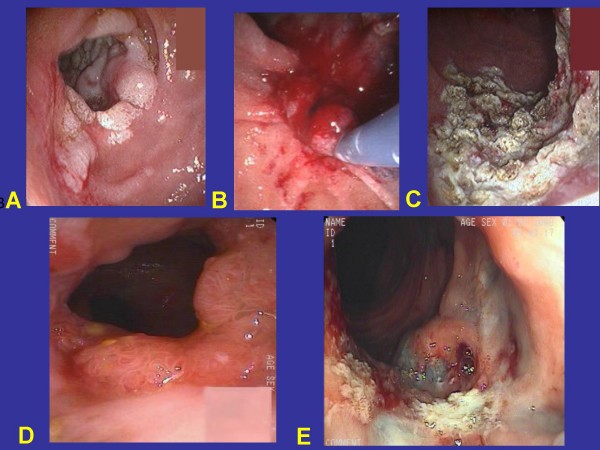
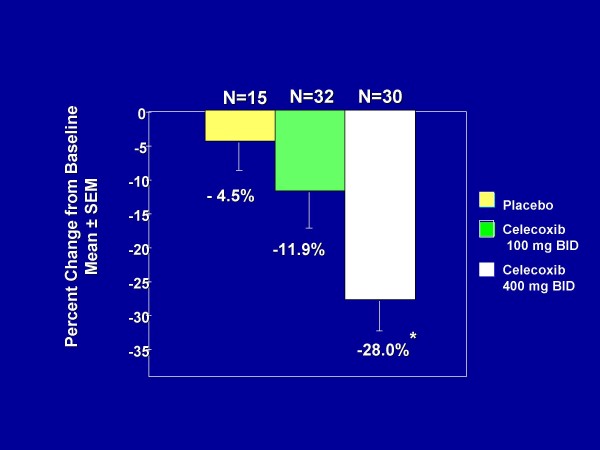
Familial adenomatous polyposis (FAP) is characterized by the development of many tens to thousands of adenomas in the rectum and colon during the second decade of life. FAP has an incidence at birth of about 1/8,300, it manifests equally in both sexes, and accounts for less than 1% of colorectal cancer (CRC) cases. In the European Union, prevalence has been estimated at 1/11,300-37,600. Most patients are asymptomatic for years until the adenomas are large and numerous, and cause rectal bleeding or even anemia, or cancer develops. Generally, cancers start to develop a decade after the appearance of the polyps. Nonspecific symptoms may include constipation or diarrhea, abdominal pain, palpable abdominal masses and weight loss. FAP may present with some extraintestinal manifestations such as osteomas, dental abnormalities (unerupted teeth, congenital absence of one or more teeth, supernumerary teeth, dentigerous cysts and odontomas), congenital hypertrophy of the retinal pigment epithelium (CHRPE), desmoid tumors, and extracolonic cancers (thyroid, liver, bile ducts and central nervous system). A less aggressive variant of FAP, attenuated FAP (AFAP), is characterized by fewer colorectal adenomatous polyps (usually 10 to 100), later age of adenoma appearance and a lower cancer risk. Some lesions (skull and mandible osteomas, dental abnormalities, and fibromas on the scalp, shoulders, arms and back) are indicative of the Gardner variant of FAP. Classic FAP is inherited in an autosomal dominant manner and results from a germline mutation in the adenomatous polyposis (APC) gene. Most patients (~70%) have a family history of colorectal polyps and cancer. In a subset of individuals, a MUTYH mutation causes a recessively inherited polyposis condition, MUTYH-associated polyposis (MAP), which is characterized by a slightly increased risk of developing CRC and polyps/adenomas in both the upper and lower gastrointestinal tract. Diagnosis is based on a suggestive family history, clinical findings, and large bowel endoscopy or full colonoscopy. Whenever possible, the clinical diagnosis should be confirmed by genetic testing. When the APC mutation in the family has been identified, genetic testing of all first-degree relatives should be performed. Presymptomatic and prenatal (amniocentesis and chorionic villous sampling), and even preimplantation genetic testing is possible. Referral to a geneticist or genetic counselor is mandatory. Differential diagnoses include other disorders causing multiple polyps (such as Peutz-Jeghers syndrome, familial juvenile polyps or hyperplastic polyposis, hereditary mixed polyposis syndromes, and Lynch syndrome). Cancer prevention and maintaining a good quality of life are the main goals of management and regular and systematic follow-up and supportive care should be offered to all patients. By the late teens or early twenties, colorectal cancer prophylactic surgery is advocated. The recommended alternatives are total proctocolectomy and ileoanal pouch or ileorectal anastomosis for AFAP. Duodenal cancer and desmoids are the two main causes of mortality after total colectomy, they need to be identified early and treated. Upper endoscopy is necessary for surveillance to reduce the risk of ampullary and duodenal cancer. Patients with progressive tumors and unresectable disease may respond or stabilize with a combination of cytotoxic chemotherapy and surgery (when possible to perform). Adjunctive therapy with celecoxib has been approved by the US Food and Drug Administration and the European Medicines Agency in patients with FAP. Individuals with FAP carry a 100% risk of CRC; however, this risk is reduced significantly when patients enter a screening-treatment program.
Figures
References
-
- Alm T. Surgical treatment of hereditary adenomatosis of the colon and rectum in Sweden during the last 20 years. Part II. Patients with prophylactic operations, primary and late results. Discussion and summary. Acta Chir Scand. 1975;141:228–237. - PubMed
-
- European Medicines Agency Doc. Ref.:EMEA/COMP/264/04draft http://www.emea.europa.eu/pdfs/human/comp/opinion/026404en.pdf
-
- Petersen GM, Slack J, Nakamura Y. Screening guidelines and premorbid diagnosis of familial adenomatous polyposis using linkage. Gastroenterology. 1991;100:1658–1664. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
