

Diagnosis of Charcot-Marie-Tooth disease
- PMID: 19826499
- PMCID: PMC2760395
- DOI: 10.1155/2009/985415
Diagnosis of Charcot-Marie-Tooth disease
Abstract
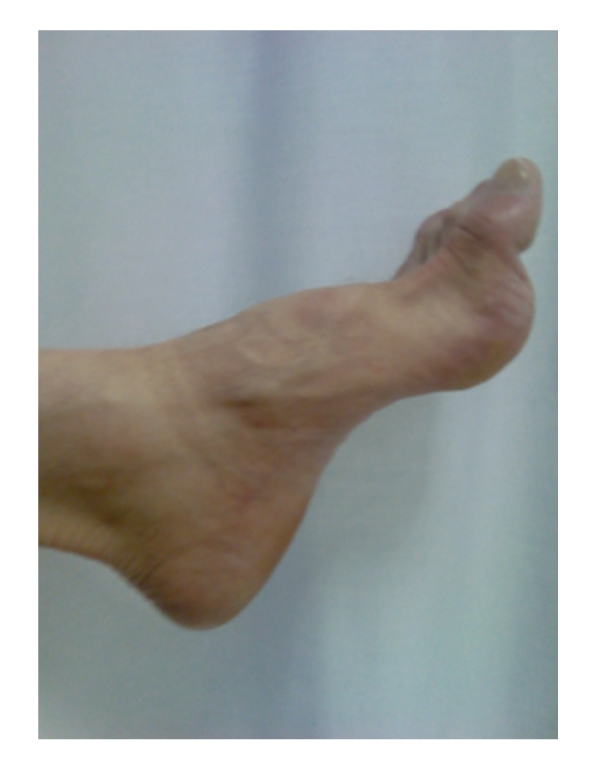
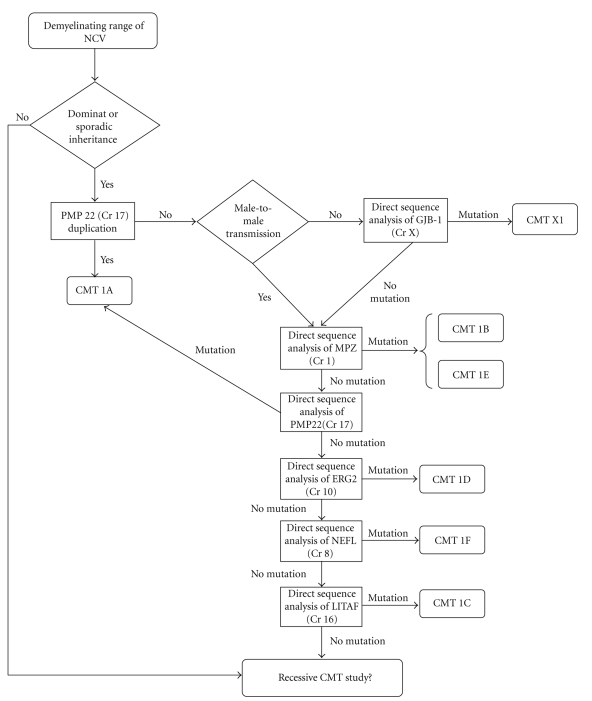
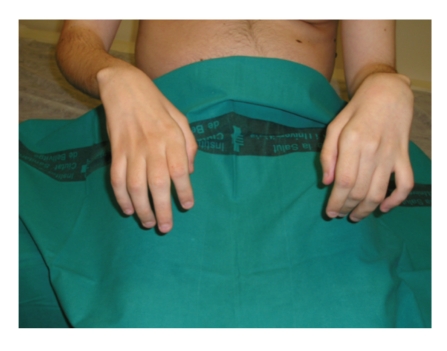
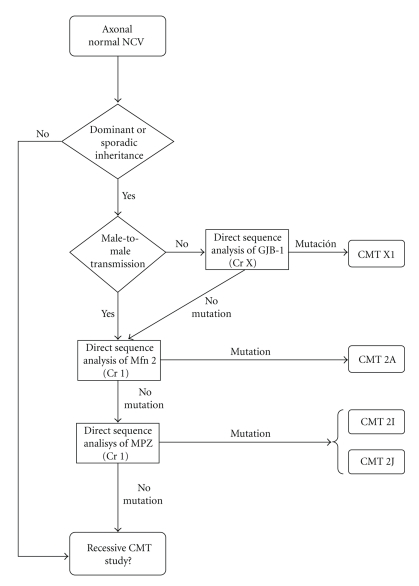
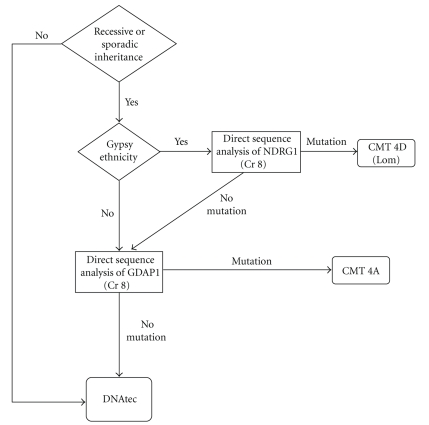
Charcot-Marie-Tooth (CMT) disease or hereditary motor and sensory neuropathy (HMSN) is a genetically heterogeneous group of conditions that affect the peripheral nervous system. The disease is characterized by degeneration or abnormal development of peripheral nerves and exhibits a range of patterns of genetic transmission. In the majority of cases, CMT first appears in infancy, and its manifestations include clumsiness of gait, predominantly distal muscular atrophy of the limbs, and deformity of the feet in the form of foot drop. It can be classified according to the pattern of transmission (autosomal dominant, autosomal recessive, or X linked), according to electrophysiological findings (demyelinating or axonal), or according to the causative mutant gene. The classification of CMT is complex and undergoes constant revision as new genes and mutations are discovered. In this paper, we review the most efficient diagnostic algorithms for the molecular diagnosis of CMT, which are based on clinical and electrophysiological data.
Figures
References
-
- Dyck P, Chance P, Lebo R, et al. Hereditary motor and sensory neuropaties. In: Dick P, editor. Peripheral Neuropathy. 3rd edition. Philadelphia, Pa, USA: W.B. Saunders; 1993. pp. 1094–1136.
-
- Sevilla T, Vilchez JJ. Diferentes fenotipos del síndrome de Charcot-Marie-Tooth causados por mutaciones del mismo gen: siguen siendo útiles los criterios de clasificación clásicos? Neurologia. 2004;19(5):264–271. - PubMed
-
- Berciano J, Combarros O. Hereditary neuropathies. Current Opinion in Neurology. 2003;16(5):613–622. - PubMed
-
- Emery AEH. Population frequencies of inherited neuromuscular diseases—a world survey. Neuromuscular Disorders. 1991;1(1):19–29. - PubMed
