

Epigenetic regulation of the honey bee transcriptome: unravelling the nature of methylated genes
- PMID: 19828049
- PMCID: PMC2768749
- DOI: 10.1186/1471-2164-10-472
Epigenetic regulation of the honey bee transcriptome: unravelling the nature of methylated genes
Abstract
Background: Epigenetic modification of DNA via methylation is one of the key inventions in eukaryotic evolution. It provides a source for the switching of gene activities, the maintenance of stable phenotypes and the integration of environmental and genomic signals. Although this process is widespread among eukaryotes, both the patterns of methylation and their relevant biological roles not only vary noticeably in different lineages, but often are poorly understood. In addition, the evolutionary origins of DNA methylation in multicellular organisms remain enigmatic. Here we used a new 'epigenetic' model, the social honey bee Apis mellifera, to gain insights into the significance of methylated genes.
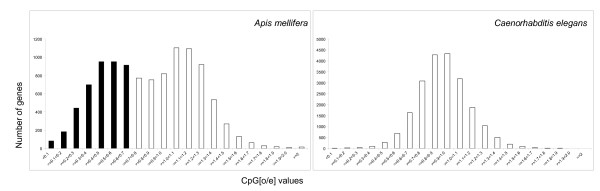
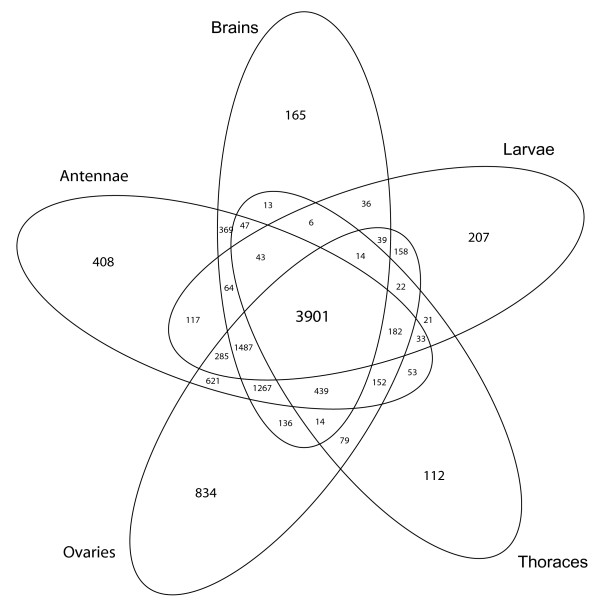
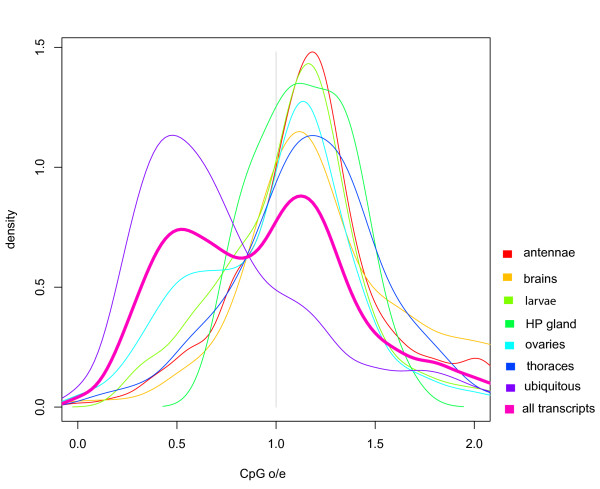
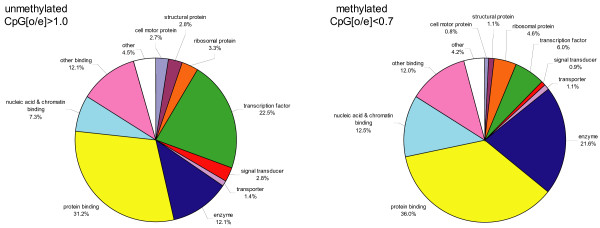
Results: We combined microarray profiling of several tissues with genome-scale bioinformatics and bisulfite sequencing of selected genes to study the honey bee methylome. We find that around 35% of the annotated honey bee genes are expected to be methylated at the CpG dinucleotides by a highly conserved DNA methylation system. We show that one unifying feature of the methylated genes in this species is their broad pattern of expression and the associated 'housekeeping' roles. In contrast, genes involved in more stringently regulated spatial or temporal functions are predicted to be un-methylated.
Conclusion: Our data suggest that honey bees use CpG methylation of intragenic regions as an epigenetic mechanism to control the levels of activity of the genes that are broadly expressed and might be needed for conserved core biological processes in virtually every type of cell. We discuss the implications of our findings for genome-scale regulatory network structures and the evolution of the role(s) of DNA methylation in eukaryotes. Our findings are particularly important in the context of the emerging evidence that environmental factors can influence the epigenetic settings of some genes and lead to serious metabolic and behavioural disorders.
Figures
Similar articles
-
The honey bee epigenomes: differential methylation of brain DNA in queens and workers.PLoS Biol. 2010 Nov 2;8(11):e1000506. doi: 10.1371/journal.pbio.1000506. PLoS Biol. 2010. PMID: 21072239 Free PMC article.
-
Roles of DNA Methylation in Color Alternation of Eastern Honey Bees (Apis cerana) Induced by the Royal Jelly of Western Honey Bees (Apis mellifera).Int J Mol Sci. 2024 Mar 16;25(6):3368. doi: 10.3390/ijms25063368. Int J Mol Sci. 2024. PMID: 38542342 Free PMC article.
-
Functional conservation of DNA methylation in the pea aphid and the honeybee.Genome Biol Evol. 2010;2:719-28. doi: 10.1093/gbe/evq057. Epub 2010 Sep 20. Genome Biol Evol. 2010. PMID: 20855427 Free PMC article.
-
DNA Methylation and Gene Regulation in Honeybees: From Genome-Wide Analyses to Obligatory Epialleles.Adv Exp Med Biol. 2016;945:193-211. doi: 10.1007/978-3-319-43624-1_9. Adv Exp Med Biol. 2016. PMID: 27826840 Review.
-
Monitoring methylation changes in cancer.Adv Biochem Eng Biotechnol. 2007;104:1-11. doi: 10.1007/10_024. Adv Biochem Eng Biotechnol. 2007. PMID: 17290816 Review.
Cited by
-
Deposition of histone variant H2A.Z within gene bodies regulates responsive genes.PLoS Genet. 2012;8(10):e1002988. doi: 10.1371/journal.pgen.1002988. Epub 2012 Oct 11. PLoS Genet. 2012. PMID: 23071449 Free PMC article.
-
DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees.Proc Natl Acad Sci U S A. 2012 Mar 27;109(13):4968-73. doi: 10.1073/pnas.1202392109. Epub 2012 Mar 13. Proc Natl Acad Sci U S A. 2012. PMID: 22416128 Free PMC article.
-
Comparative analyses of DNA methylation and sequence evolution using Nasonia genomes.Mol Biol Evol. 2011 Dec;28(12):3345-54. doi: 10.1093/molbev/msr168. Epub 2011 Jun 20. Mol Biol Evol. 2011. PMID: 21693438 Free PMC article.
-
Hemimetabolous genomes reveal molecular basis of termite eusociality.Nat Ecol Evol. 2018 Mar;2(3):557-566. doi: 10.1038/s41559-017-0459-1. Epub 2018 Feb 5. Nat Ecol Evol. 2018. PMID: 29403074 Free PMC article.
-
The genomic impact of 100 million years of social evolution in seven ant species.Trends Genet. 2012 Jan;28(1):14-21. doi: 10.1016/j.tig.2011.08.005. Epub 2011 Oct 5. Trends Genet. 2012. PMID: 21982512 Free PMC article. Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
