

Data recovery and integration from public databases uncovers transformation-specific transcriptional downregulation of cAMP-PKA pathway-encoding genes
- PMID: 19828069
- PMCID: PMC2762058
- DOI: 10.1186/1471-2105-10-S12-S1
Data recovery and integration from public databases uncovers transformation-specific transcriptional downregulation of cAMP-PKA pathway-encoding genes
Abstract
Background: The integration of data from multiple genome-wide assays is essential for understanding dynamic spatio-temporal interactions within cells. Such integration, which leads to a more complete view of cellular processes, offers the opportunity to rationalize better the high amount of "omics" data freely available in several public databases.In particular, integration of microarray-derived transcriptome data with other high-throughput analyses (genomic and mutational analysis, promoter analysis) may allow us to unravel transcriptional regulatory networks under a variety of physio-pathological situations, such as the alteration in the cross-talk between signal transduction pathways in transformed cells.
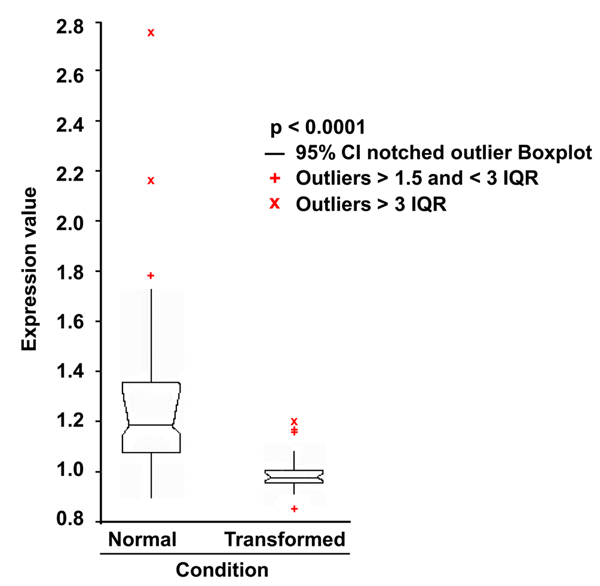
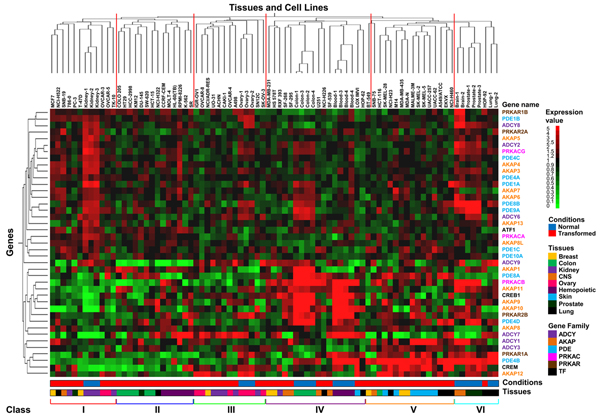
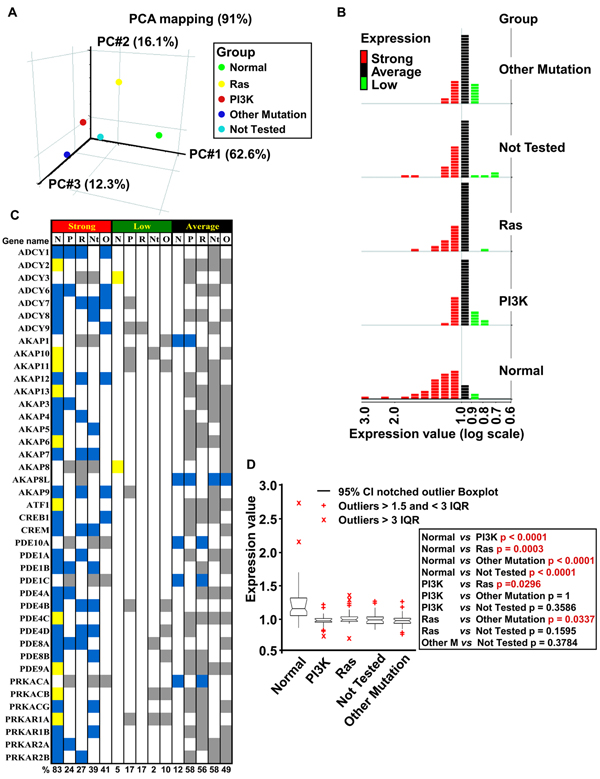
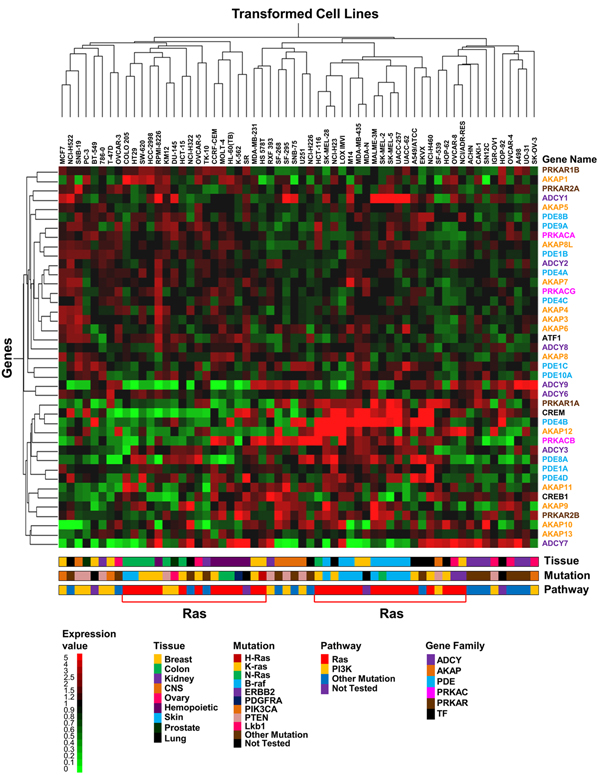
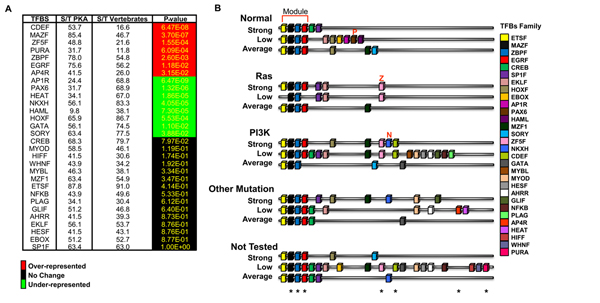
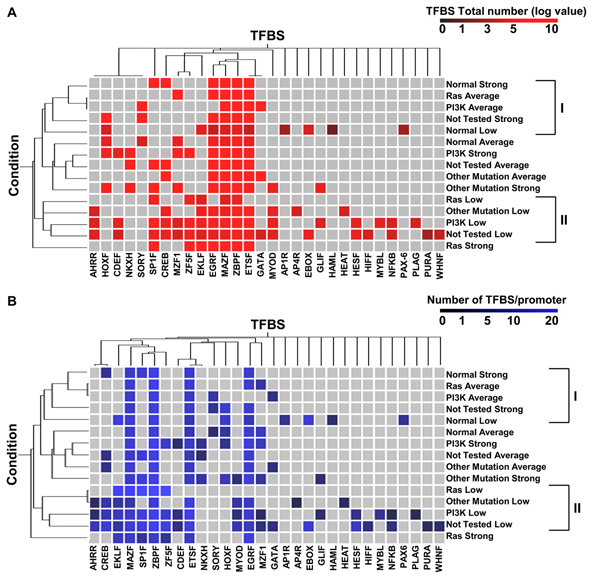
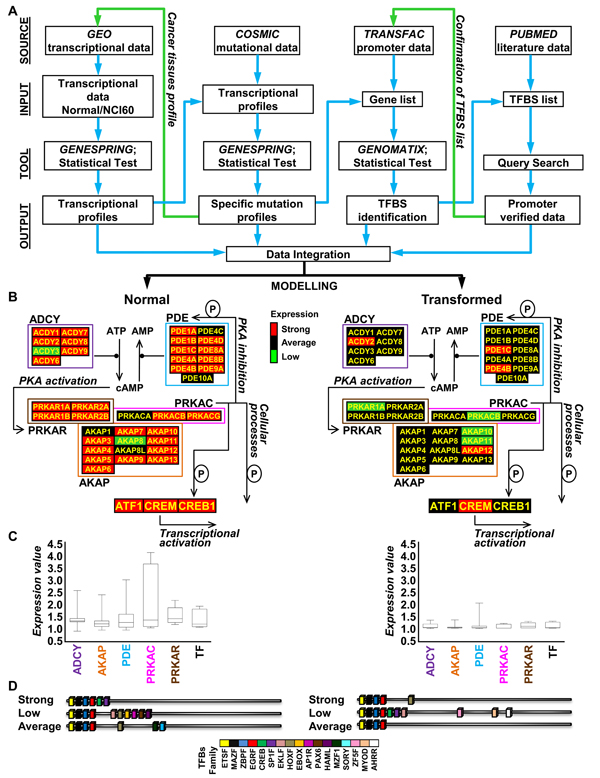
Results: Here we sequentially apply web-based and statistical tools to a case study: the role of oncogenic activation of different signal transduction pathways in the transcriptional regulation of genes encoding proteins involved in the cAMP-PKA pathway. To this end, we first re-analyzed available genome-wide expression data for genes encoding proteins of the downstream branch of the PKA pathway in normal tissues and human tumor cell lines. Then, in order to identify mutation-dependent transcriptional signatures, we classified cancer cells as a function of their mutational state. The results of such procedure were used as a starting point to analyze the structure of PKA pathway-encoding genes promoters, leading to identification of specific combinations of transcription factor binding sites, which are neatly consistent with available experimental data and help to clarify the relation between gene expression, transcriptional factors and oncogenes in our case study.
Conclusions: Genome-wide, large-scale "omics" experimental technologies give different, complementary perspectives on the structure and regulatory properties of complex systems. Even the relatively simple, integrated workflow presented here offers opportunities not only for filtering data noise intrinsic in high throughput data, but also to progressively extract novel information that would have remained hidden otherwise. In fact we have been able to detect a strong transcriptional repression of genes encoding proteins of cAMP/PKA pathway in cancer cells of different genetic origins. The basic workflow presented herein may be easily extended by incorporating other tools and can be applied even by researchers with poor bioinformatics skills.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
