

A Perl procedure for protein identification by Peptide Mass Fingerprinting
- PMID: 19828071
- PMCID: PMC2762060
- DOI: 10.1186/1471-2105-10-S12-S11
A Perl procedure for protein identification by Peptide Mass Fingerprinting
Abstract
Background: One of the topics of major interest in proteomics is protein identification. Protein identification can be achieved by analyzing the mass spectrum of a protein sample through different approaches. One of them, called Peptide Mass Fingerprinting (PMF), combines mass spectrometry (MS) data with searching strategies in a suitable database of known protein to provide a list of candidate proteins ranked by a score. To this aim, several algorithms and software tools have been proposed. However, the scoring methods and mainly the statistical evaluation of the results can be significantly improved.
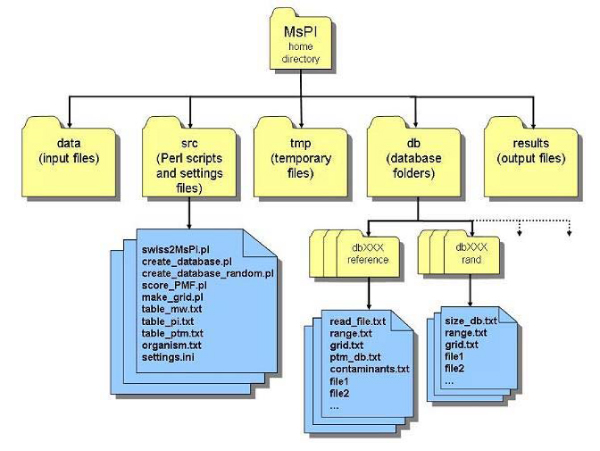
Results: In this work, a Perl procedure for protein identification by PMF, called MsPI (Mass spectrometry Protein Identification), is presented. The implemented scoring methods were derived from the literature. MsPI implements a strategy to remove the contaminant masses present in the acquired spectra. Moreover, MsPI includes a statistical method to assign to each candidate protein, in addition to the scoring value, a p-value. Results obtained by MsPI on a dataset of 10 protein samples were compared with those achieved using two other software tools, i.e. Piums and Mascot. Piums implements one of the scoring methods available in MsPI, while Mascot is one of the most frequently used software tools in the protein identification field. MsPI scripts are available for downloading on the web site http://aimed11.unipv.it/MsPI.
Conclusion: The performances of MsPI seem to be better than those of Piums and Mascot. In fact, on the considered dataset, MsPI includes in its candidate proteins list, the "true" proteins nine times over ten, whereas Piums includes in its list the "true" proteins only four time over ten. Even if Mascot also correctly includes in the candidates list the "true" proteins nine times over ten, it provides longer candidate lists, therefore increasing the number of false positives when the molecular weight of the proteins in the sample is approximatively known (e.g. by the 1-D/2-D electrophoresis gel). Moreover, being MsPI a Perl tool, it can be easily extended and customized by the final users.
Figures

References
-
- Downard K. Mass Spectrometry: A Foundation Course. Royal Society of Chemistry; 2004.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
