

TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration
- PMID: 19833869
- PMCID: PMC2762420
- DOI: 10.1073/pnas.0908767106
TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration
Abstract
Frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS) are neurodegenerative diseases that show considerable clinical and pathologic overlap, with no effective treatments available. Mutations in the RNA binding protein TDP-43 were recently identified in patients with familial amyotrophic lateral sclerosis (ALS), and TDP-43 aggregates are found in both ALS and FTLD-U (FTLD with ubiquitin aggregates), suggesting a common underlying mechanism. We report that mice expressing a mutant form of human TDP-43 develop a progressive and fatal neurodegenerative disease reminiscent of both ALS and FTLD-U. Despite universal transgene expression throughout the nervous system, pathologic aggregates of ubiquitinated proteins accumulate only in specific neuronal populations, including layer 5 pyramidal neurons in frontal cortex, as well as spinal motor neurons, recapitulating the phenomenon of selective vulnerability seen in patients with FTLD-U and ALS. Surprisingly, cytoplasmic TDP-43 aggregates are not present, and hence are not required for TDP-43-induced neurodegeneration. These results indicate that the cellular and molecular substrates for selective vulnerability in FTLD-U and ALS are shared between mice and humans, and suggest that altered DNA/RNA-binding protein function, rather than toxic aggregation, is central to TDP-43-related neurodegeneration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
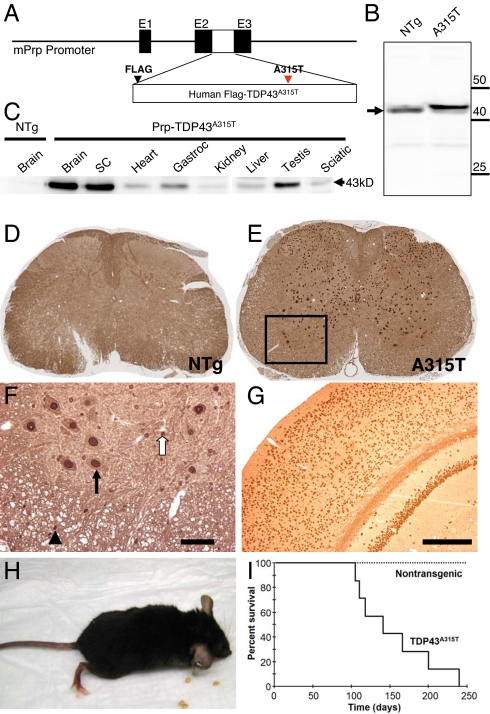
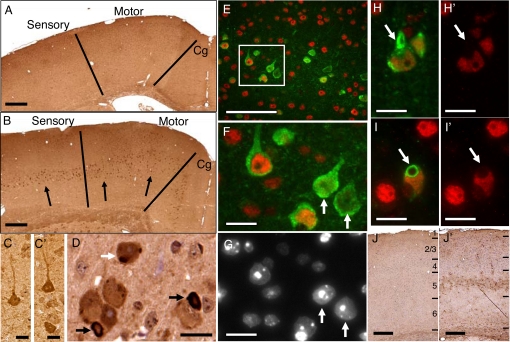
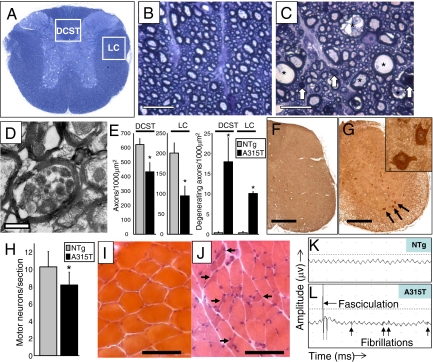
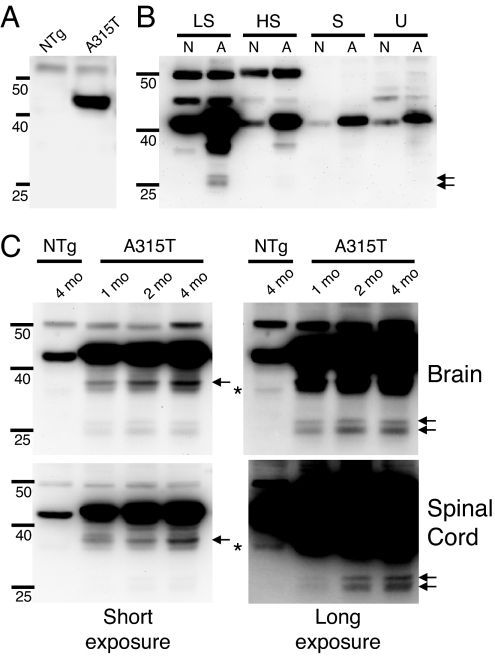
References
-
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. - PubMed
-
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat Rev Neurosci. 2006;7:710–723. - PubMed
-
- Murphy JM, et al. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:530–534. - PubMed
-
- Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
