

Limiting the persistence of a chromosome break diminishes its mutagenic potential
- PMID: 19834534
- PMCID: PMC2752804
- DOI: 10.1371/journal.pgen.1000683
Limiting the persistence of a chromosome break diminishes its mutagenic potential
Abstract
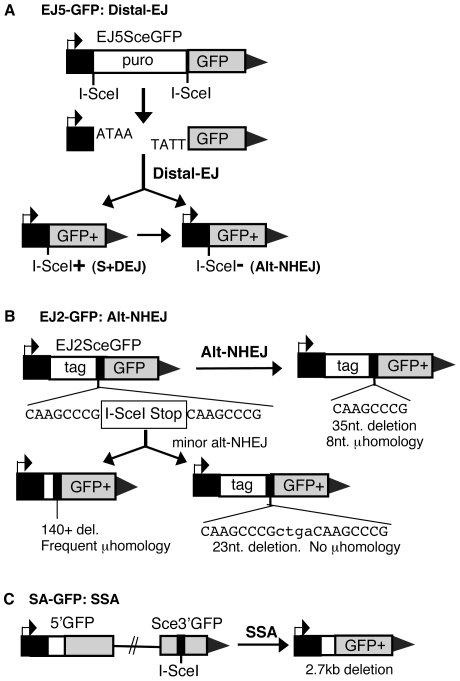
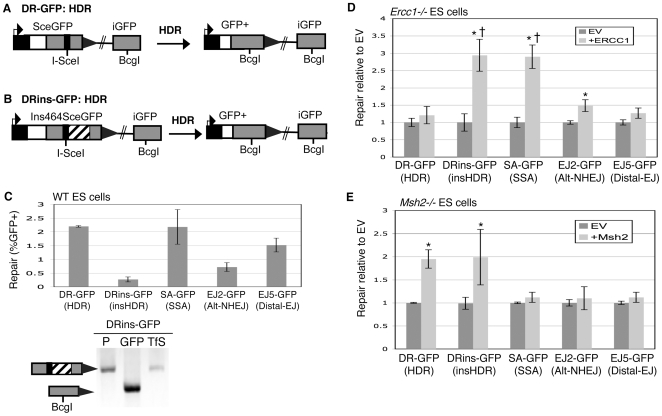
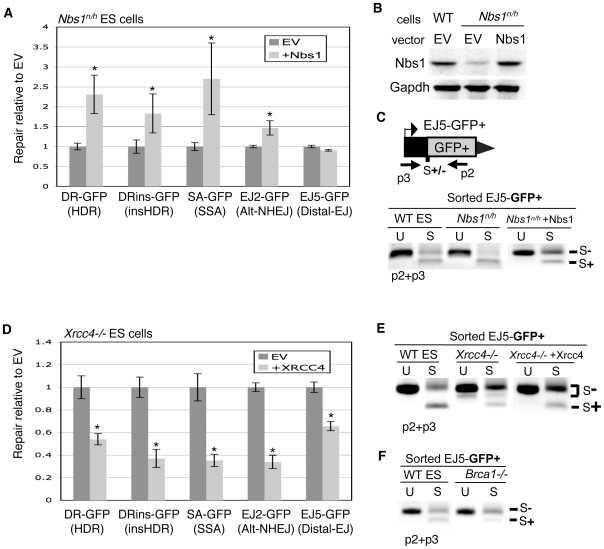
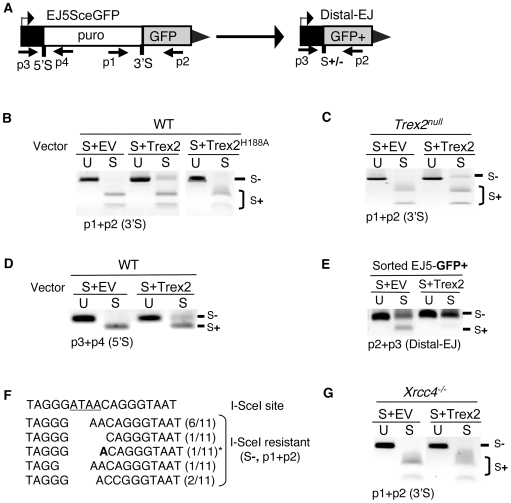
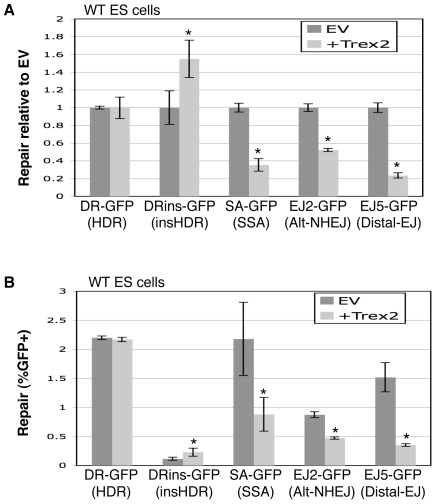
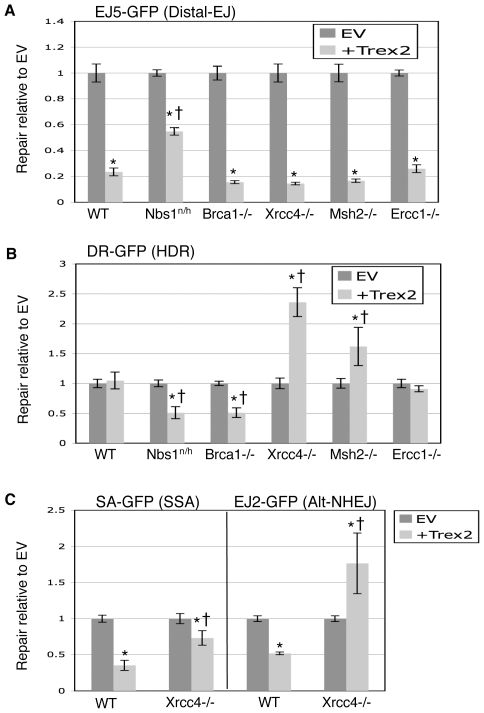
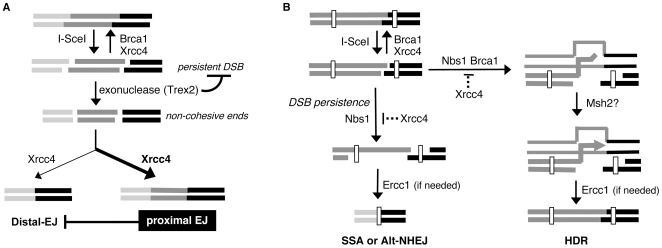
To characterize the repair pathways of chromosome double-strand breaks (DSBs), one approach involves monitoring the repair of site-specific DSBs generated by rare-cutting endonucleases, such as I-SceI. Using this method, we first describe the roles of Ercc1, Msh2, Nbs1, Xrcc4, and Brca1 in a set of distinct repair events. Subsequently, we considered that the outcome of such assays could be influenced by the persistent nature of I-SceI-induced DSBs, in that end-joining (EJ) products that restore the I-SceI site are prone to repeated cutting. To address this aspect of repair, we modified I-SceI-induced DSBs by co-expressing I-SceI with a non-processive 3' exonuclease, Trex2, which we predicted would cause partial degradation of I-SceI 3' overhangs. We find that Trex2 expression facilitates the formation of I-SceI-resistant EJ products, which reduces the potential for repeated cutting by I-SceI and, hence, limits the persistence of I-SceI-induced DSBs. Using this approach, we find that Trex2 expression causes a significant reduction in the frequency of repair pathways that result in substantial deletion mutations: EJ between distal ends of two tandem DSBs, single-strand annealing, and alternative-NHEJ. In contrast, Trex2 expression does not inhibit homology-directed repair. These results indicate that limiting the persistence of a DSB causes a reduction in the frequency of repair pathways that lead to significant genetic loss. Furthermore, we find that individual genetic factors play distinct roles during repair of non-cohesive DSB ends that are generated via co-expression of I-SceI with Trex2.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Wyman C, Kanaar R. DNA double-strand break repair: all's well that ends well. Annu Rev Genet. 2006;40:363–383. - PubMed
-
- Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003;3:639–649. - PubMed
-
- Ma Y, Lu H, Schwarz K, Lieber MR. Repair of double-strand DNA breaks by the human nonhomologous DNA end joining pathway: the iterative processing model. Cell Cycle. 2005;4:1193–1200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
