

doi: 10.1186/gb-2009-10-10-r115.
Epub 2009 Oct 16.
Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts
Affiliations
- PMID: 19835606
- PMCID: PMC2784330
- DOI: 10.1186/gb-2009-10-10-r115
Item in Clipboard
Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts
Genome Biol.
2009.
Abstract
Targeted RNA-Seq combines next-generation sequencing with capture of sequences from a relevant subset of a transcriptome. When testing by capturing sequences from a tumor cDNA library by hybridization to oligonucleotide probes specific for 467 cancer-related genes, this method showed high selectivity, improved mutation detection enabling discovery of novel chimeric transcripts, and provided RNA expression data. Thus, targeted RNA-Seq produces an enhanced view of the molecular state of a set of "high interest" genes.
Figures
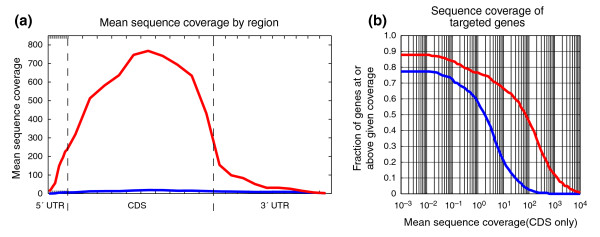
Increase in sequence coverage. (a) Mean sequence coverage by region. Transcript regions (5' UTR, CDS, 3' UTR) were divided into deciles, and the sequence coverage for each decile was averaged across all 887 target transcripts. Coverage is displayed for before hybrid selection (blue) and after hybrid selection (red). The average length of each region is 292, 2,136, and 1,729, respectively. (b) Distribution of sequence coverage for targeted genes. For each sequence-coverage threshold (x-axis), the fraction of 467 genes at or above that threshold is plotted (y-axis) for before hybrid selection (blue) and after hybrid selection (red).
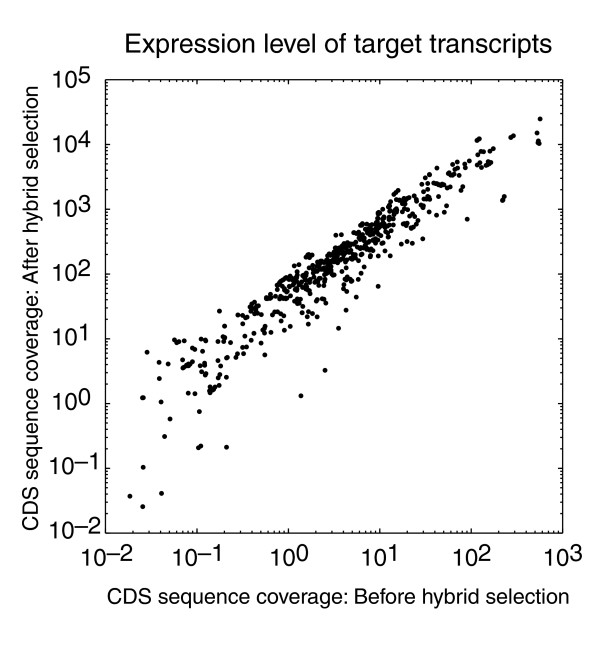
Preservation of expression levels of target transcripts in hybrid selection. For each target transcript, the sequence coverage of the coding region is plotted before and after hybrid selection.

Sequences from NUP214-XKR3 fusion transcripts detected after hybrid selection. After hybrid selection, 152 reads were aligned to the transcriptome and detected as NUP214-XKR3 fusions. From top to bottom, we observed 137, four, eight, and three reads for these transcripts. The NUP214 (exon 27) to XKR3 (exon 4) has a stop codon downstream (not shown). Only NUP214 (exon 29) to XKR3 (exon 4) retains an open reading frame downstream of the fusion. Before hybrid selection, eight reads were aligned to the transcriptome and detected as NUP214-XKR3 fusions; only the NUP214 (exon 29) to XKR3 (exon 2) transcript was detected. Sequence from NUP214 DNA is shown as lower case, and from XKR3, as bold and upper case.
References
-
- Zhao Q, Caballero OL, Levy S, Stevenson BJ, Iseli C, de Souza SJ, Galante PA, Busam D, Leversha MA, Chadalavada K, Rogers YH, Venter JC, Simpson AJ, Strausberg RL. Transcriptome-guided characterization of genomic rearrangements in a breast cancer cell line. Proc Natl Acad Sci USA. 2009;106:1886–1891. doi: 10.1073/pnas.0812945106. - DOI - PMC - PubMed
-
- McLendon R, Friedman A, Bigner D, Van Meir EG, Brat D, Mastrogianakis GM, Olson JJ, Mikkelsen T, Lehman N, Aldape K, Yung WK, Bogler O, Weinstein JN, VandenBerg S, Berger M, Prados M, Muzny D, Morgan M, Scherer S, Sabo A, Nazareth L, Lewis L, Hall O, Zhu Y, Ren Y, Alvi O, Yao J, Hawes A, Jhangiani S, Fowler G. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
