

Discovery through the computational microscope
- PMID: 19836330
- PMCID: PMC2927212
- DOI: 10.1016/j.str.2009.09.001
Discovery through the computational microscope
Abstract
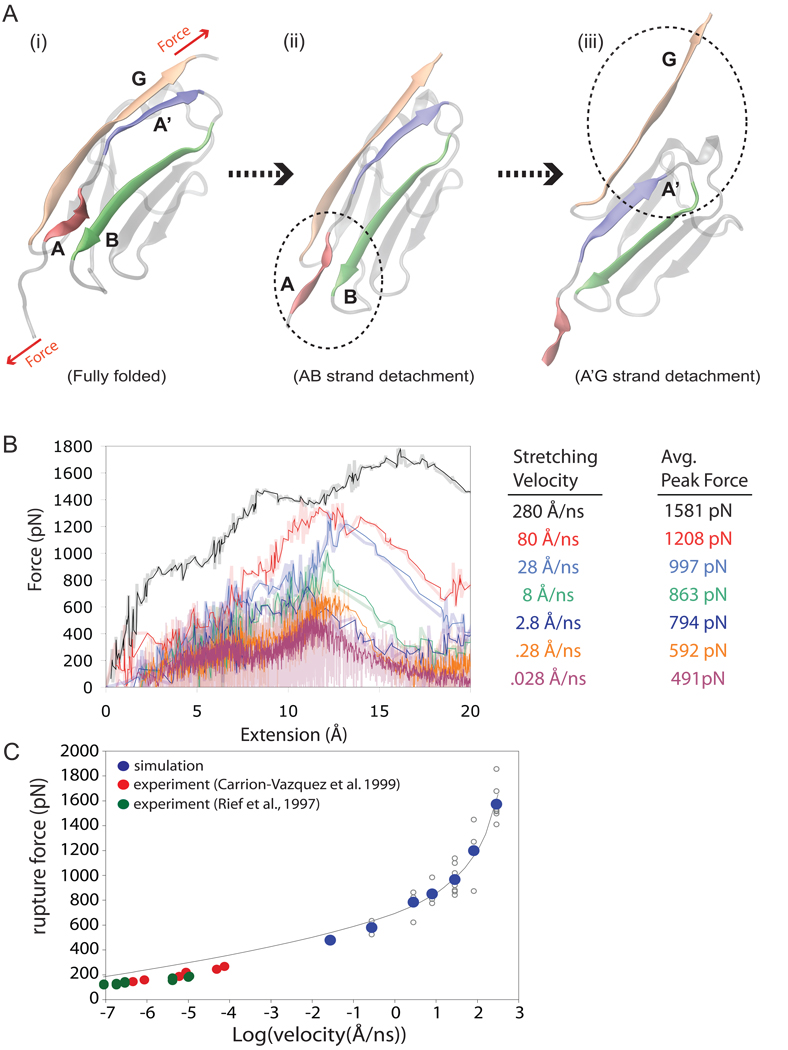
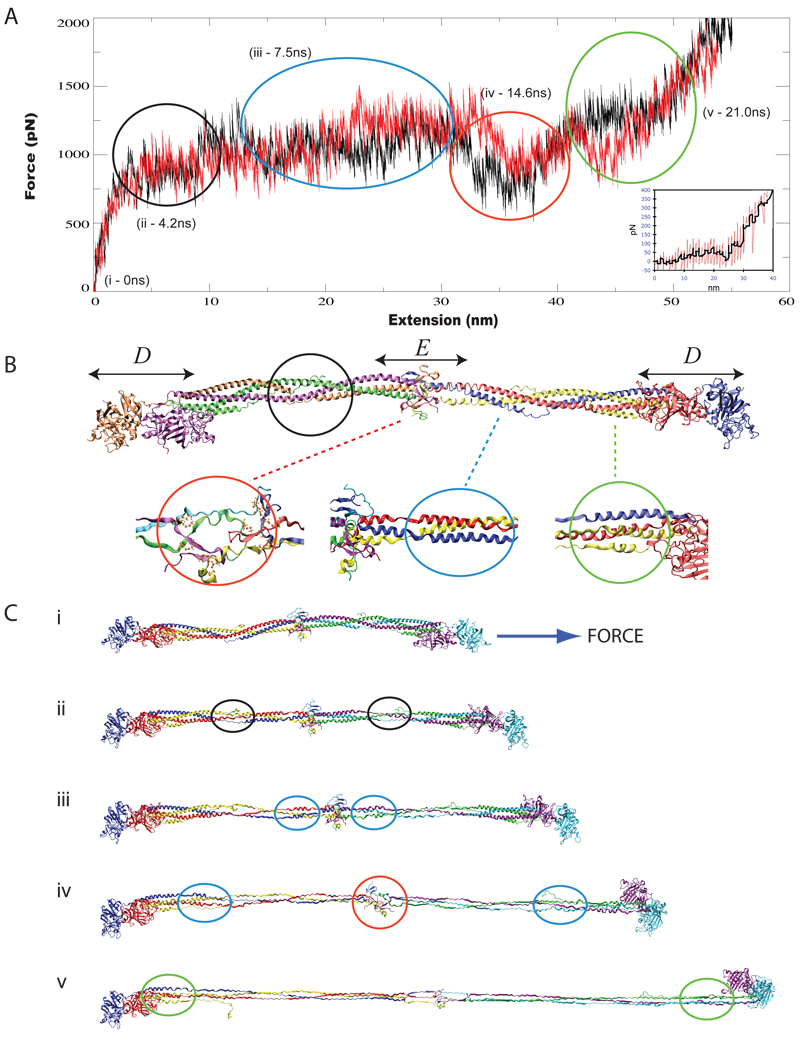
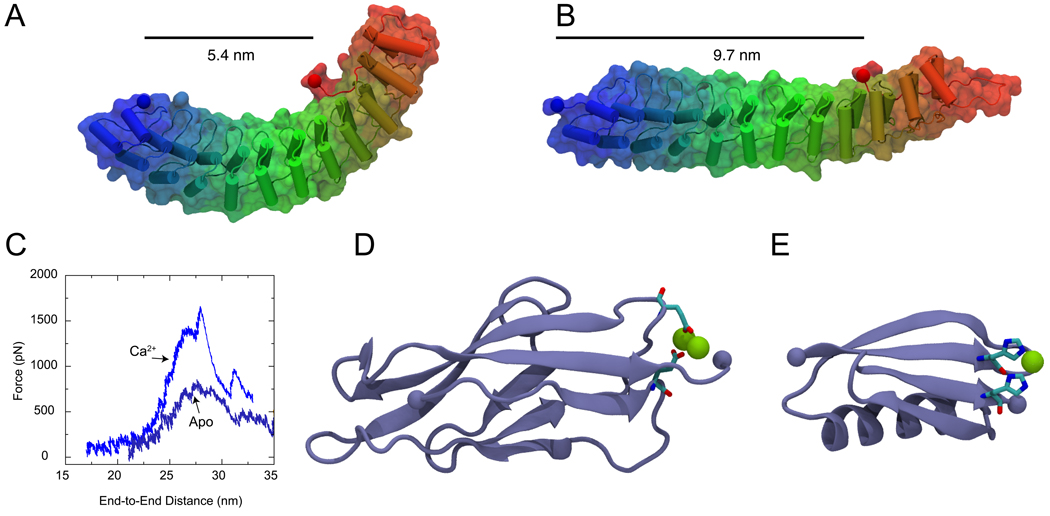
All-atom molecular dynamics simulations have become increasingly popular as a tool to investigate protein function and dynamics. However, researchers are concerned about the short time scales covered by simulations, the apparent impossibility to model large and integral biomolecular systems, and the actual predictive power of the molecular dynamics methodology. Here we review simulations that were in the past both hotly disputed and considered key successes, namely of proteins with mainly mechanical functions (titin, fibrinogen, ankyrin, and cadherin). The simulation work covered shows how state-of-the-art modeling alleviates some of the prior concerns and how unrefuted discoveries are made through the "computational microscope."
Figures
References
-
- Aksimentiev A, Brunner R, Cohen J, Comer J, Cruz-Chu E, Hardy D, Rajan A, Shih A, Sigalov G, Yin Y, et al. Protocols in Nanostructure Design. Methods in Molecular Biology. Humana Press; 2008. Computer modeling in biotechnology, a partner in development; pp. 181–234. - PubMed
-
- Arkhipov A, Freddolino PL, Schulten K. Stability and dynamics of virus capsids described by coarse-grained modeling. Structure. 2006;14:1767–1777. - PubMed
-
- Arkin IT, Xu H, Jensen M, Arbely E, Bennett ER, Bowers KJ, Chow E, Dror RO, Eastwood MP, Flitman-Tene R, et al. Mechanism of Na+/H+ antiporting. Science. 2007;317:799–803. - PubMed
-
- Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, et al. The complete gene sequence of titin, expression of an unusual approximate to 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circulation Res. 2001;89:1065–1072. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
