

Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa
- PMID: 19836337
- PMCID: PMC2775066
- DOI: 10.1016/j.str.2009.08.010
Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa
Abstract
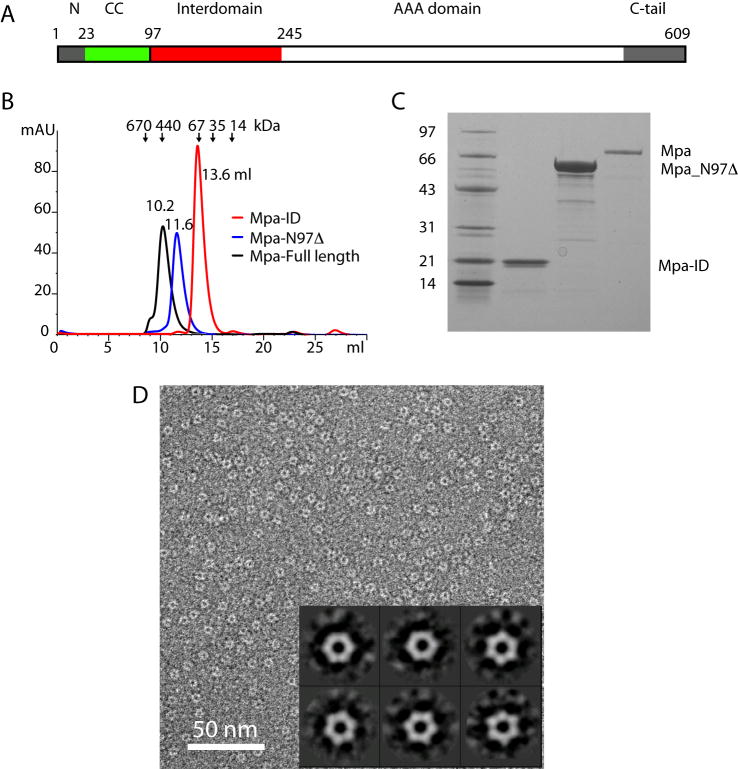
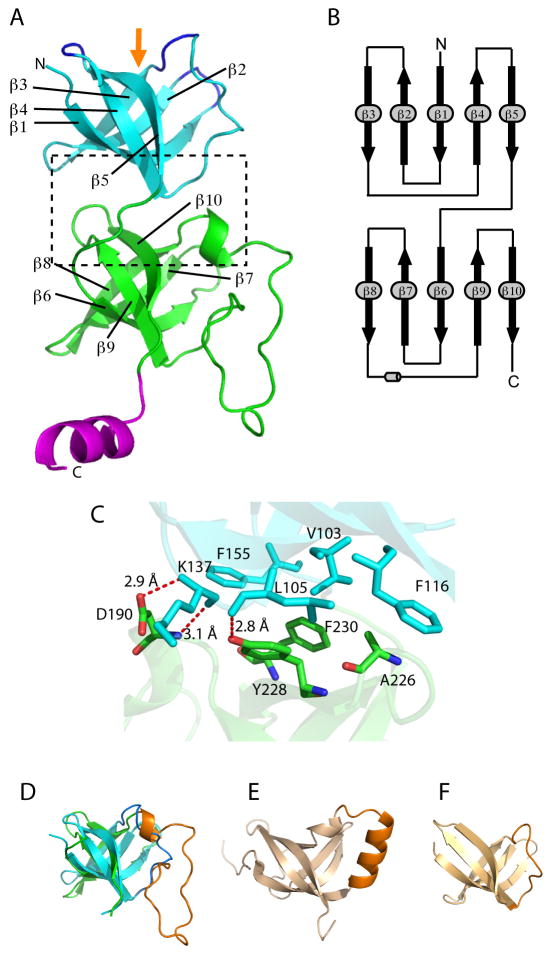
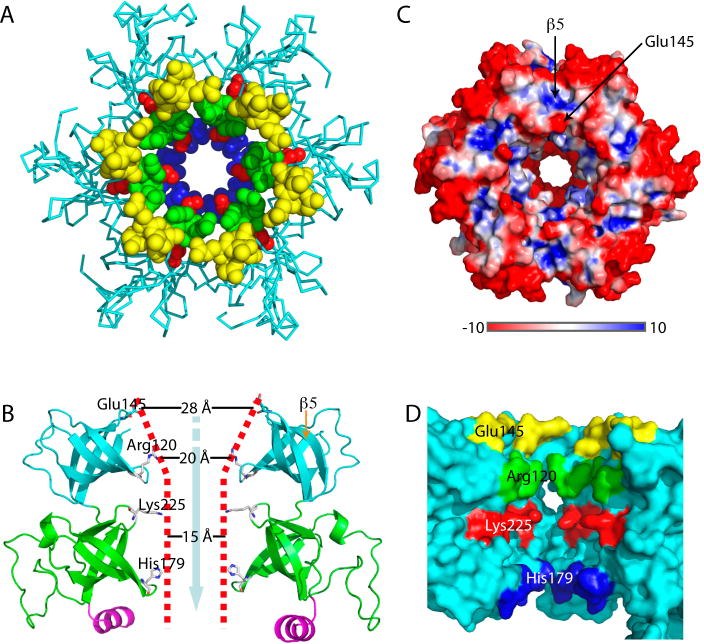
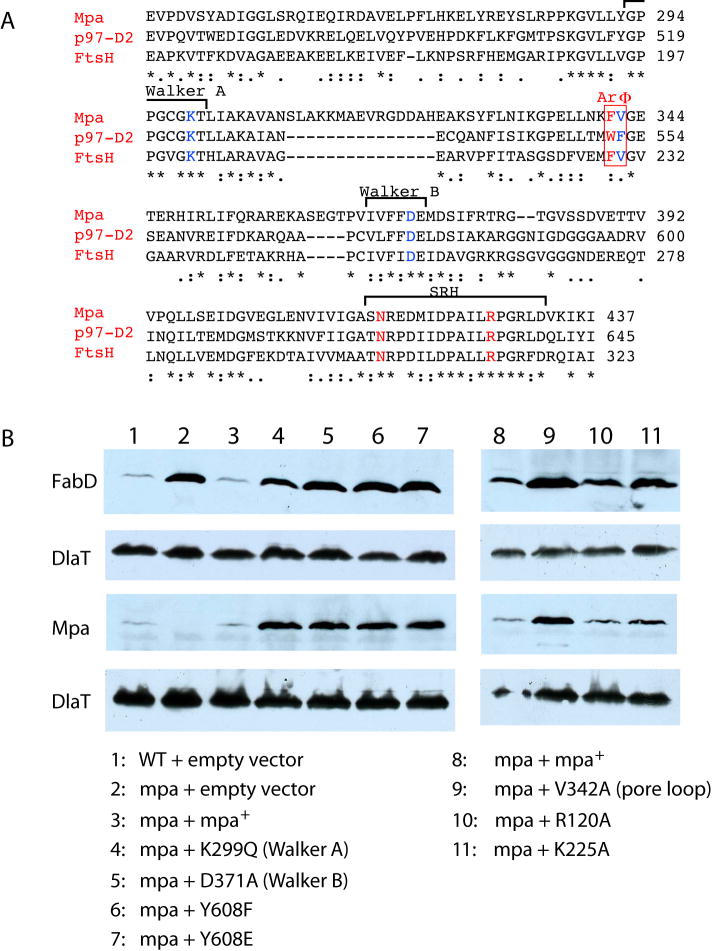
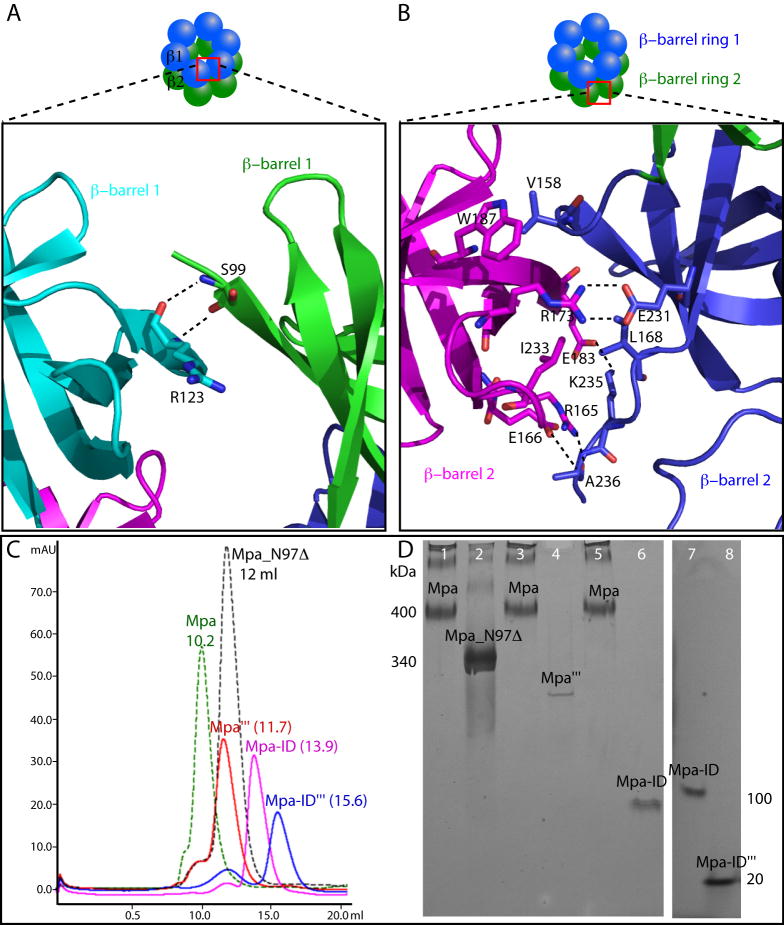
Proteasome-mediated protein turnover in all domains of life is an energy-dependent process that requires ATPase activity. Mycobacterium tuberculosis (Mtb) was recently shown to possess a ubiquitin-like proteasome pathway that plays an essential role in Mtb resistance to killing by products of host macrophages. Here we report our structural and biochemical investigation of Mpa, the presumptive Mtb proteasomal ATPase. We demonstrate that Mpa binds to the Mtb proteasome in the presence of ATPgammaS, providing the physical evidence that Mpa is the proteasomal ATPase. X-ray crystallographic determination of the conserved interdomain showed a five stranded double beta barrel structure containing a Greek key motif. Structure and mutational analysis indicate a major role of the interdomain for Mpa hexamerization. Our mutational and functional studies further suggest that the central channel in the Mpa hexamer is involved in protein substrate translocation and degradation. These studies provide insights into how a bacterial proteasomal ATPase interacts with and facilitates protein degradation by the proteasome.
Figures

Comment in
-
How ATPases unravel a mystery.Structure. 2009 Oct 14;17(10):1279-81. doi: 10.1016/j.str.2009.09.003. Structure. 2009. PMID: 19836328
References
-
- Alexandrescu AT, Gittis AG, Abeygunawardana C, Shortle D. NMR structure of a stable “OB-fold” sub-domain isolated from staphylococcal nuclease. J Mol Biol. 1995;250:134–143. - PubMed
-
- Alexandrescu AT, Jaravine VA, Dames SA, Lamour FP. NMR hydrogen exchange of the OB-fold protein LysN as a function of denaturant: the most conserved elements of structure are the most stable to unfolding. J Mol Biol. 1999;289:1041–1054. - PubMed
-
- Benaroudj N, Zwickl P, Seemuller E, Baumeister W, Goldberg AL. ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol Cell. 2003;11:69–78. - PubMed
-
- Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007;107:687–717. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
