

Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses
- PMID: 19837981
- PMCID: PMC2830836
- DOI: 10.1074/mcp.M900223-MCP200
Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses
Abstract
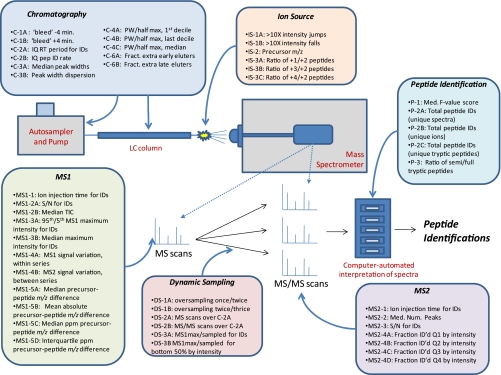
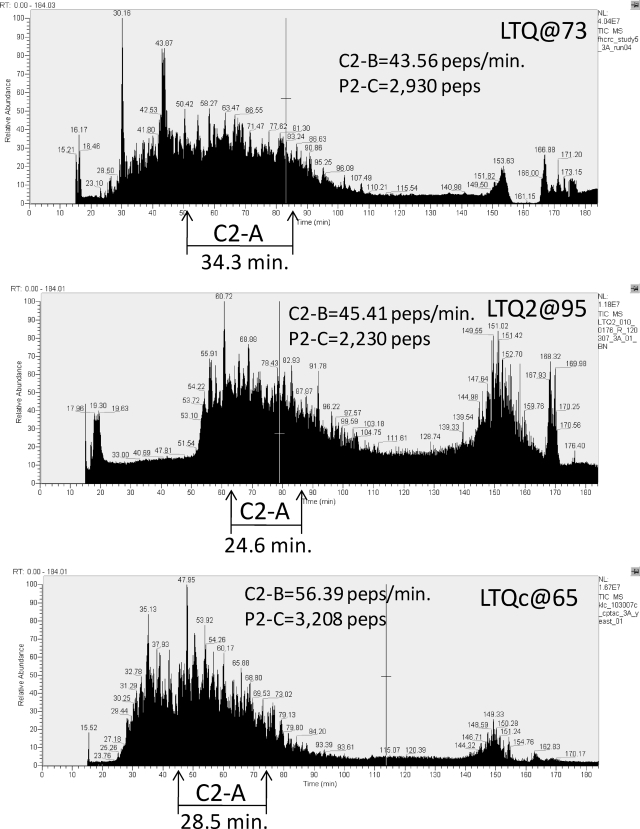
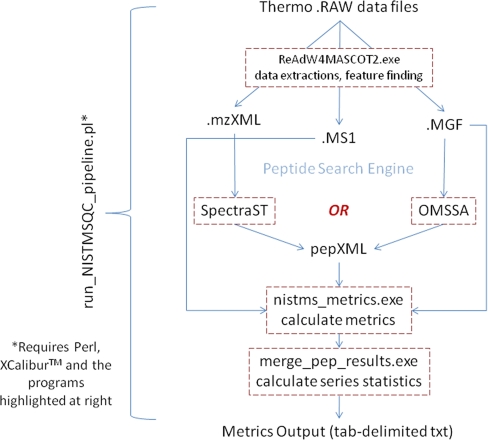
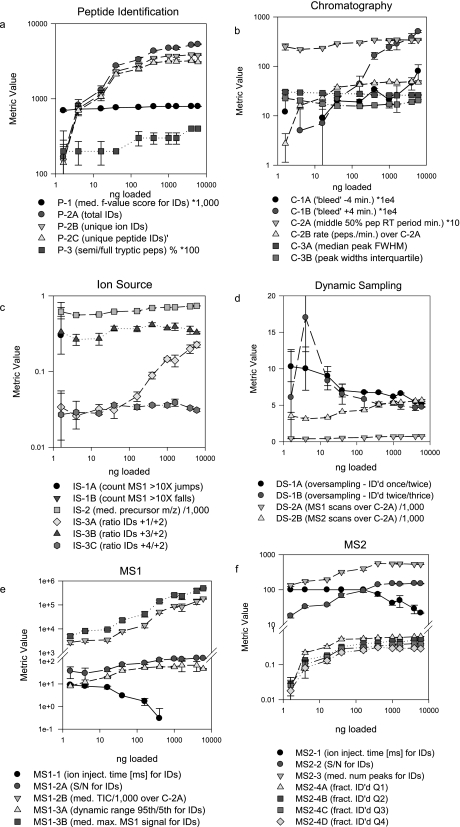
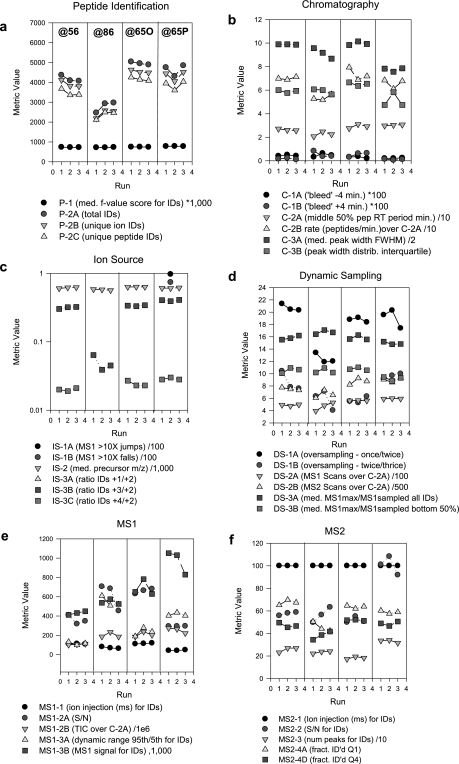
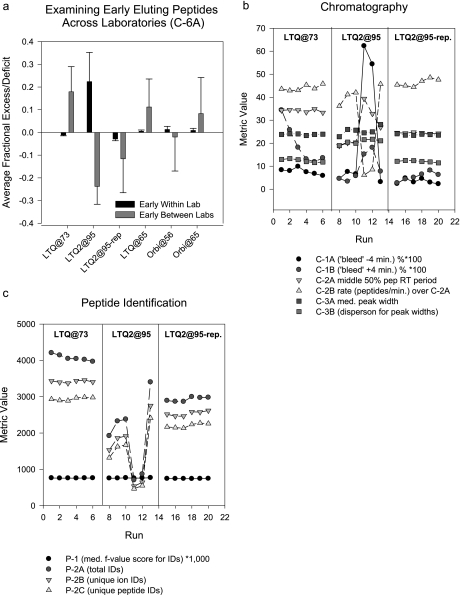
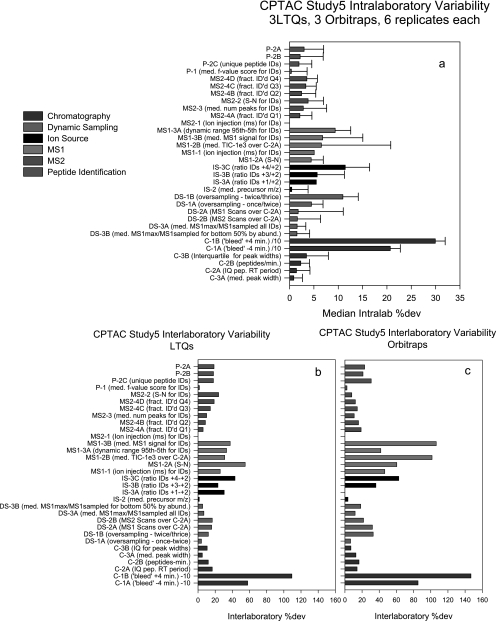
A major unmet need in LC-MS/MS-based proteomics analyses is a set of tools for quantitative assessment of system performance and evaluation of technical variability. Here we describe 46 system performance metrics for monitoring chromatographic performance, electrospray source stability, MS1 and MS2 signals, dynamic sampling of ions for MS/MS, and peptide identification. Applied to data sets from replicate LC-MS/MS analyses, these metrics displayed consistent, reasonable responses to controlled perturbations. The metrics typically displayed variations less than 10% and thus can reveal even subtle differences in performance of system components. Analyses of data from interlaboratory studies conducted under a common standard operating procedure identified outlier data and provided clues to specific causes. Moreover, interlaboratory variation reflected by the metrics indicates which system components vary the most between laboratories. Application of these metrics enables rational, quantitative quality assessment for proteomics and other LC-MS/MS analytical applications.
Figures
References
-
- Yates J. R., 3rd ( 2004) Mass spectral analysis in proteomics. Annu. Rev. Biophys. Biomol. Struct 33, 297– 316 - PubMed
-
- Aebersold R., Mann M. ( 2003) Mass spectrometry-based proteomics. Nature 422, 198– 207 - PubMed
-
- Liu H., Lin D., Yates J. R., 3rd ( 2002) Multidimensional separations for protein/peptide analysis in the post-genomic era. BioTechniques 32, 898, 900,, 902 - PubMed
-
- Eriksson J., Fenyö D. ( 2007) Improving the success rate of proteome analysis by modeling protein-abundance distributions and experimental designs. Nat. Biotechnol 25, 651– 655 - PubMed
-
- Whiteaker J. R., Zhang H., Zhao L., Wang P., Kelly-Spratt K. S., Ivey R. G., Piening B. D., Feng L. C., Kasarda E., Gurley K. E., Eng J. K., Chodosh L. A., Kemp C. J., McIntosh M. W., Paulovich A. G. ( 2007) Integrated pipeline for mass spectrometry-based discovery and confirmation of biomarkers demonstrated in a mouse model of breast cancer. J. Proteome Res 6, 3962– 3975 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
