

Visual proteomics of the human pathogen Leptospira interrogans
- PMID: 19838170
- PMCID: PMC2862215
- DOI: 10.1038/nmeth.1390
Visual proteomics of the human pathogen Leptospira interrogans
Abstract
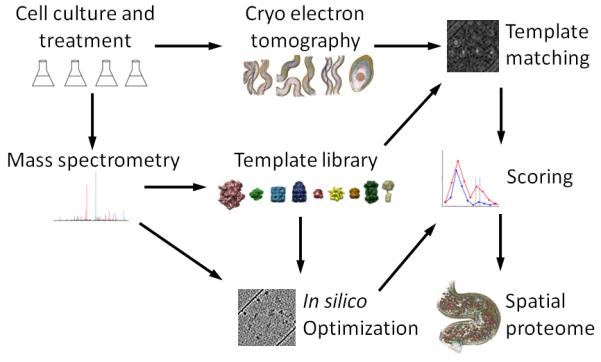
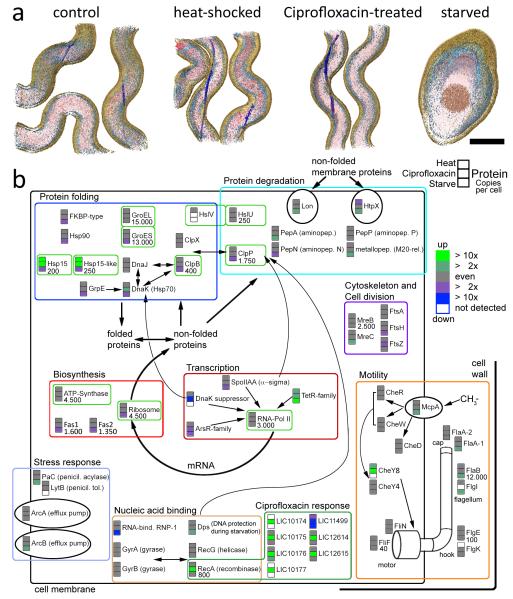
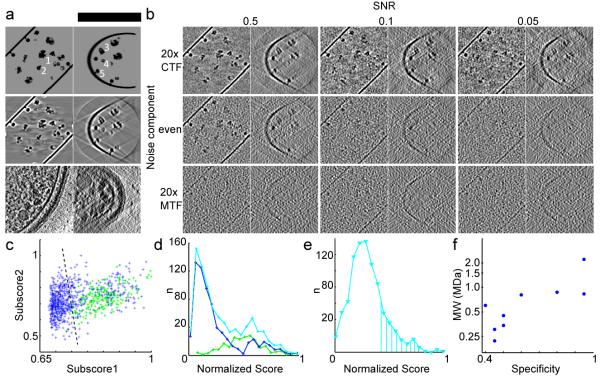
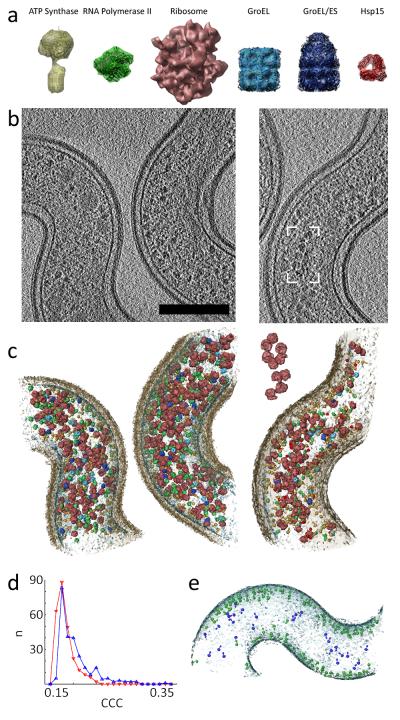
Systems biology conceptualizes biological systems as dynamic networks of interacting elements, whereby functionally important properties are thought to emerge from the structure of such networks. Owing to the ubiquitous role of complexes of interacting proteins in biological systems, their subunit composition and temporal and spatial arrangement within the cell are of particular interest. 'Visual proteomics' attempts to localize individual macromolecular complexes inside of intact cells by template matching reference structures into cryo-electron tomograms. Here we combined quantitative mass spectrometry and cryo-electron tomography to detect, count and localize specific protein complexes in the cytoplasm of the human pathogen Leptospira interrogans. We describe a scoring function for visual proteomics and assess its performance and accuracy under realistic conditions. We discuss current and general limitations of the approach, as well as expected improvements in the future.
Figures
Comment in
-
Adding a spatial dimension to the proteome.Nat Methods. 2009 Nov;6(11):798-800. doi: 10.1038/nmeth1109-798. Nat Methods. 2009. PMID: 19876016 No abstract available.
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Lucic V, Forster F, Baumeister W. Structural studies by electron tomography: from cells to molecules. Annual review of biochemistry. 2005;74:833–865. - PubMed
-
- Best C, Nickell S, Baumeister W. Localization of protein complexes by pattern recognition. Methods in cell biology. 2007;79:615–638. - PubMed
-
- Nickell S, Kofler C, Leis AP, Baumeister W. A visual approach to proteomics. Nature Reviews Molecular Cell Biology. 2006;7:225–230. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
