

Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants
- PMID: 19841300
- PMCID: PMC3025752
- DOI: 10.1161/CIRCULATIONAHA.109.863076
Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants
Abstract
Background: Genetic testing for long-QT syndrome (LQTS) has diagnostic, prognostic, and therapeutic implications. Hundreds of causative mutations in 12 known LQTS-susceptibility genes have been identified. Genetic testing that includes the 3 most commonly mutated genes is available clinically. Distinguishing pathogenic mutations from innocuous rare variants is critical to the interpretation of test results. We sought to quantify the value of mutation type and gene/protein region in determining the probability of pathogenicity for mutations.
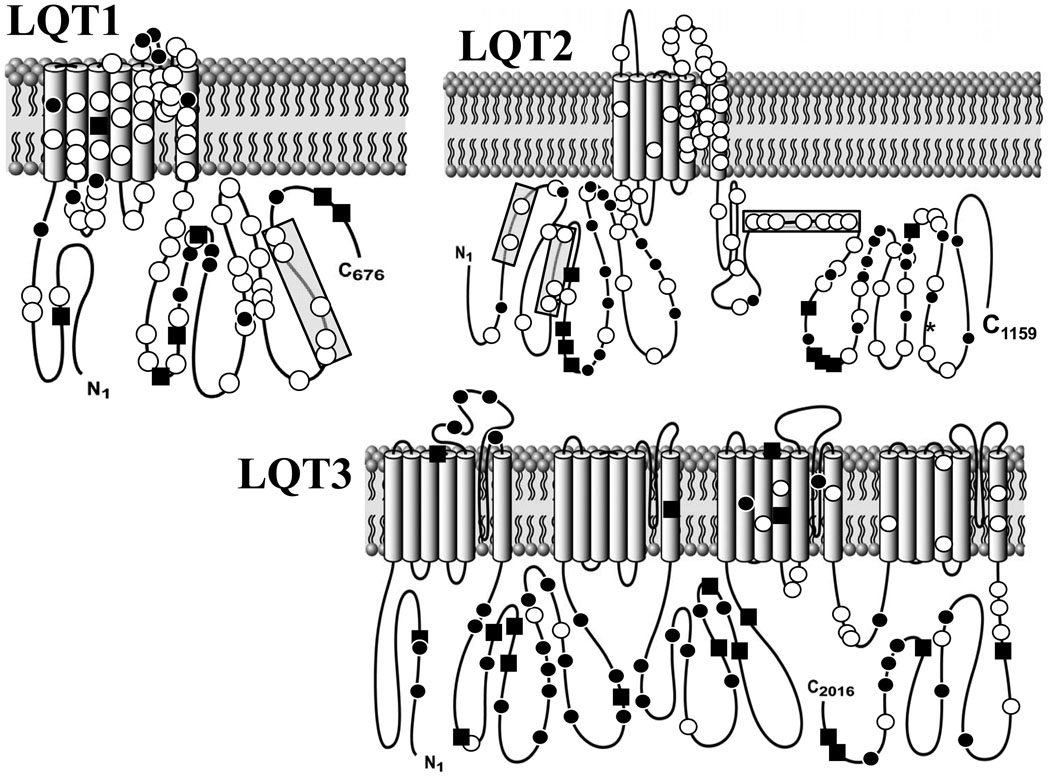
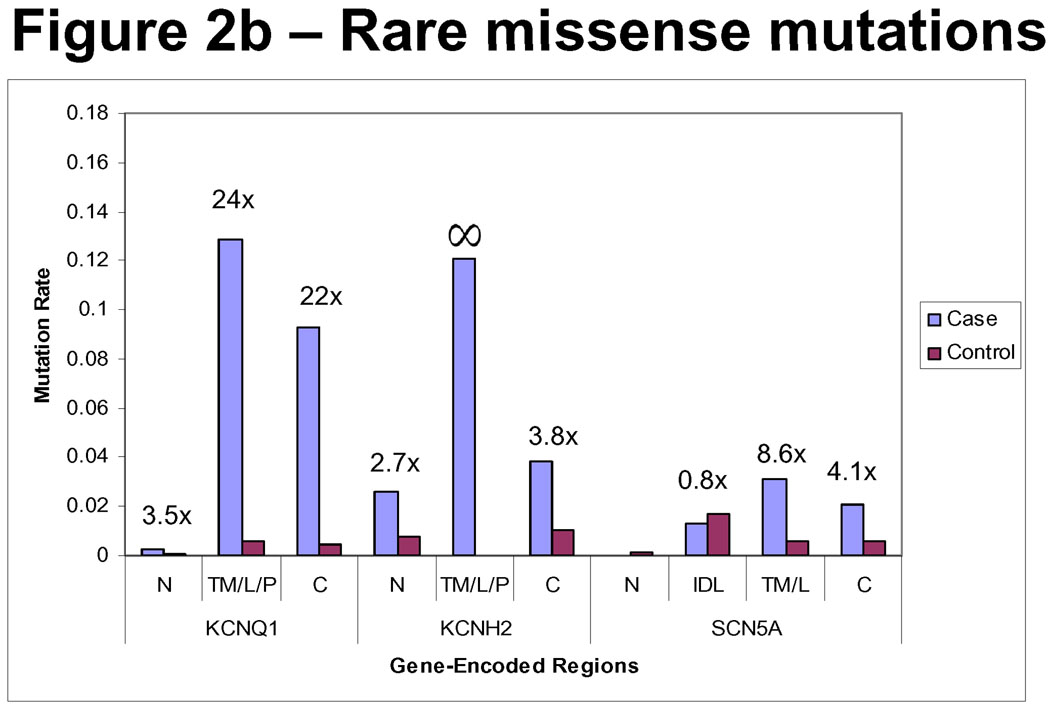
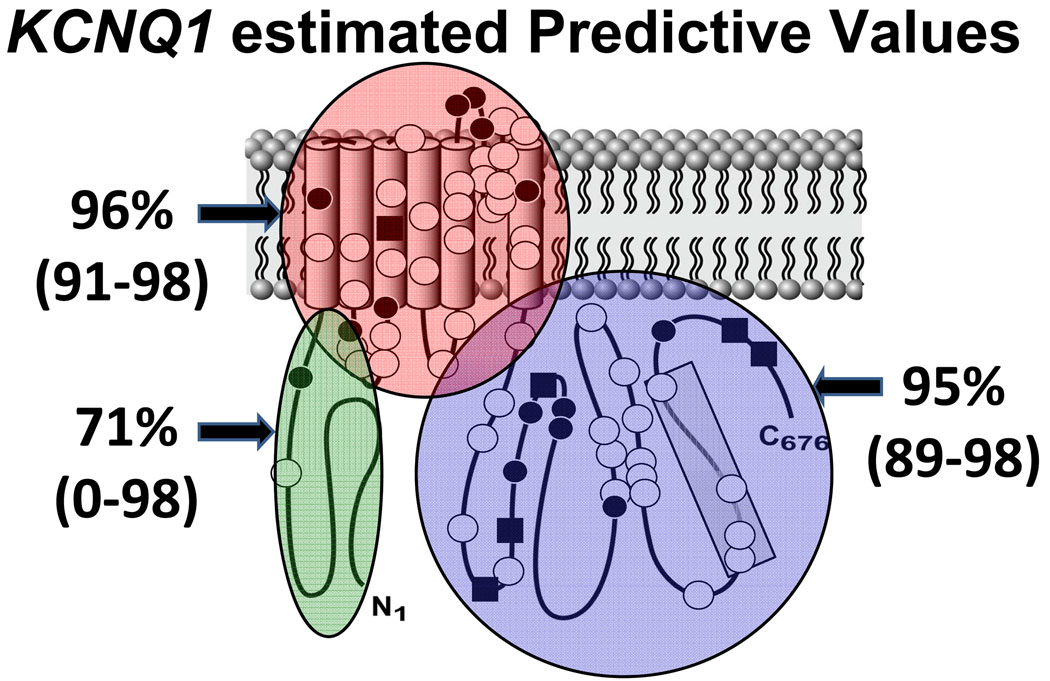
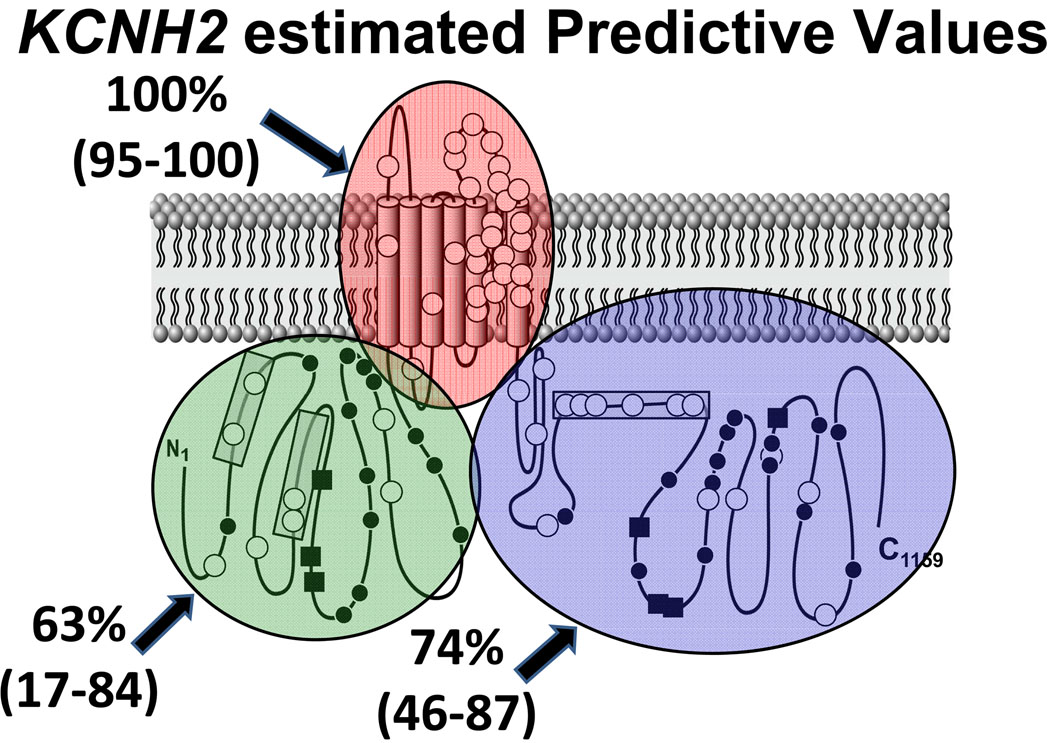
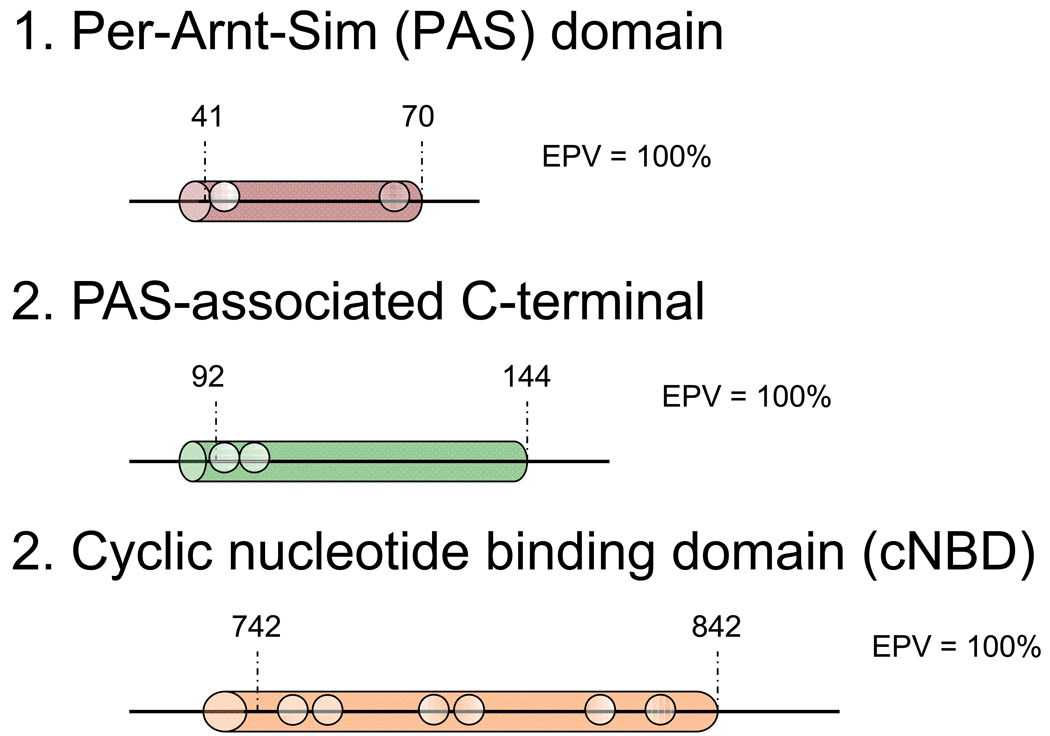
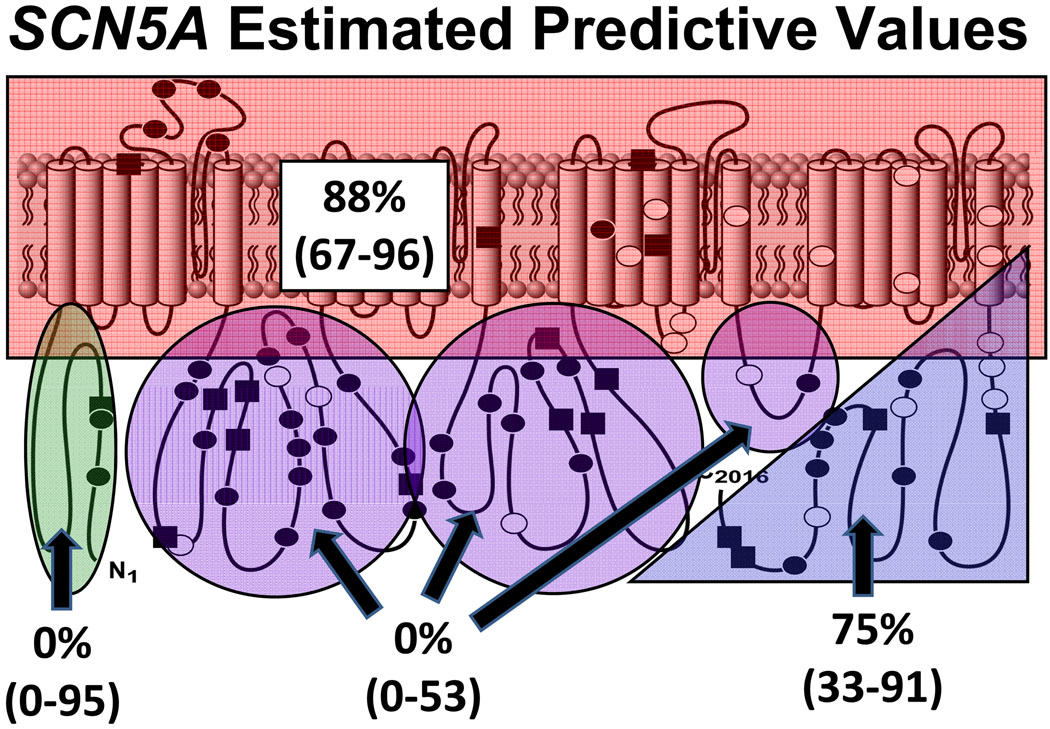
Methods and results: Type, frequency, and location of mutations across KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) were compared between 388 unrelated "definite" (clinical diagnostic score >or=4 and/or QTc >or=480 ms) cases of LQTS and >1300 healthy controls for each gene. From these data, estimated predictive values (percent of mutations found in definite cases that would cause LQTS) were determined according to mutation type and location. Mutations were 10 times more common in cases than controls (0.58 per case versus 0.06 per control). Missense mutations were the most common, accounting for 78%, 67%, and 89% of mutations in KCNQ1, KCNH2, and SCN5A in cases and >95% in controls. Nonmissense mutations have an estimated predictive value >99% regardless of location. In contrast, location appears to be critical for characterizing missense mutations. Relative frequency of missense mutations between cases and controls ranged from approximately 1:1 in the SCN5A interdomain linker to infinity in the pore, transmembrane, and linker in KCNH2. These correspond to estimated predictive values ranging from 0% in the interdomain linker of SCN5A to 100% in the transmembrane/linker/pore regions of KCNH2. The estimated predictive value is also high in the linker, pore, transmembrane, and C terminus of KCNQ1 and the transmembrane/linker of SCN5A.
Conclusions: Distinguishing pathogenic mutations from rare variants is of critical importance in the interpretation of genetic testing in LQTS. Mutation type, mutation location, and ethnic-specific
Background: should be viewed as variants of uncertain significance and prompt further investigation to clarify the likelihood of disease causation. However, mutations in regions such as the transmembrane, linker, and pore of KCNQ1 and KCNH2 may be defined confidently as high-probability LQTS-causing mutations. These findings will have implications for other genetic disorders involving mutational analysis.
Conflict of interest statement
SK: None. DJT: None. BAS and CHK are employees of PGxHealth, which offers the FAMILION® LQTS Test, and stockholders of the parent company, Clinical Data.
MSP: None. MA: None. AAMW: None. MJA is a consultant for PGxHealth and chairs their FAMILION Medical/Scientific Advisory Board (approved by Mayo Clinic’s Medical-Industry Relations Office and Conflict of Interests Review Board). In addition, “cardiac channel gene screen” and “know-how relating to long QT genetic testing” license agreements, resulting in consideration and royalty payments, were established between Genaissance Pharmaceuticals (now PGxHealth) and Mayo Medical Ventures (now Mayo Clinic Health Solutions) in 2004.
Figures


Comment in
-
Closer look at genetic testing in long-QT syndrome: will DNA diagnostics ever be enough?Circulation. 2009 Nov 3;120(18):1745-8. doi: 10.1161/CIRCULATIONAHA.109.900415. Epub 2009 Oct 19. Circulation. 2009. PMID: 19841296 No abstract available.
References
-
- GeneTests: Medical Genetics Informational Resource (database online) Seattle: University of Washington; 2007. pp. 1995–2007. Copyright.
-
- Hunter DJ, Hunter DJ, Drazen JM. Letting the genome out of the bottle--will we get our wish? N Engl J Med. 2008;358:105–107. - PubMed
-
- Shimizu W. The long QT syndrome: therapeutic implications of a genetic diagnosis. Cardiovasc Res. 2005;67:347–356. - PubMed
-
- Keating M, Atkinson D, Dunn C, Timothy K, Vincent GM, Leppert M. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science. 1991;252:704–706. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
