

Glutathione transferases are structural and functional outliers in the thioredoxin fold
- PMID: 19842715
- PMCID: PMC2778357
- DOI: 10.1021/bi901180v
Glutathione transferases are structural and functional outliers in the thioredoxin fold
Abstract
Glutathione transferases (GSTs) are ubiquitous scavengers of toxic compounds that fall, structurally and functionally, within the thioredoxin fold suprafamily. The fundamental catalytic capability of GSTs is catalysis of the nucleophilic addition or substitution of glutathione at electrophilic centers in a wide range of small electrophilic compounds. While specific GSTs have been studied in detail, little else is known about the structural and functional relationships between different groupings of GSTs. Through a global analysis of sequence and structural similarity, it was determined that variation in the binding of glutathione between the two major subgroups of cytosolic (soluble) GSTs results in a different mode of glutathione activation. Additionally, the convergent features of glutathione binding between cytosolic GSTs and mitochondrial GST kappa are described. The identification of these structural and functional themes helps to illuminate some of the fundamental contributions of the thioredoxin fold to catalysis in the GSTs and clarify how the thioredoxin fold can be modified to enable new functions.
Figures
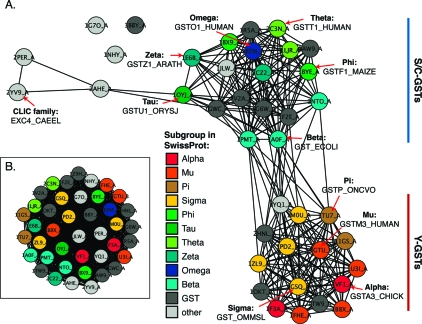
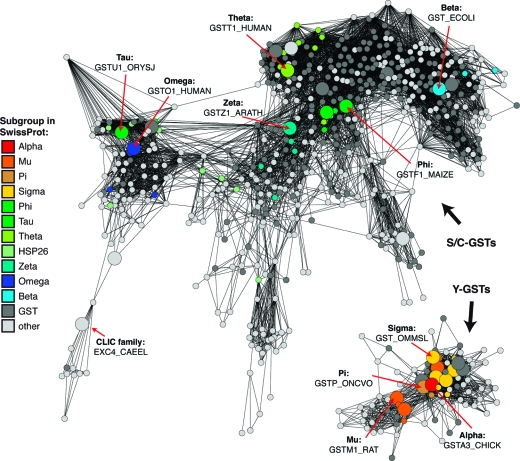
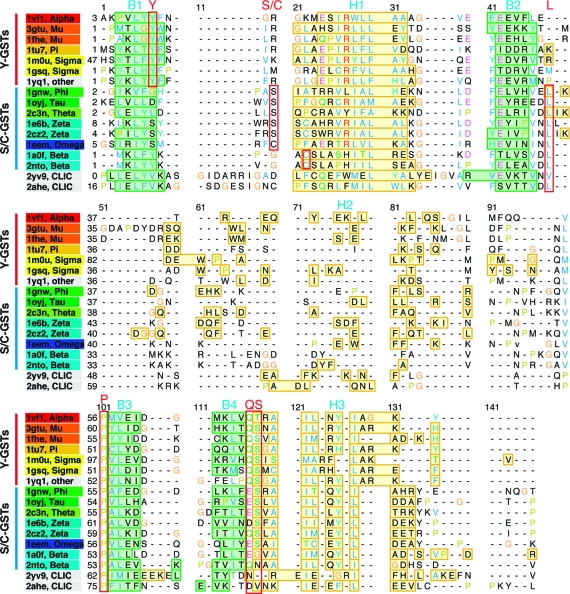
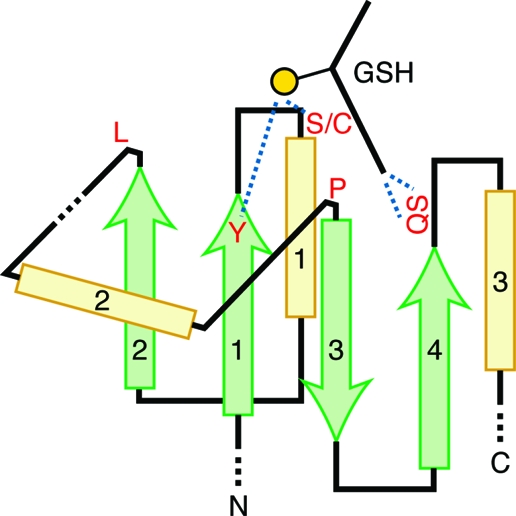
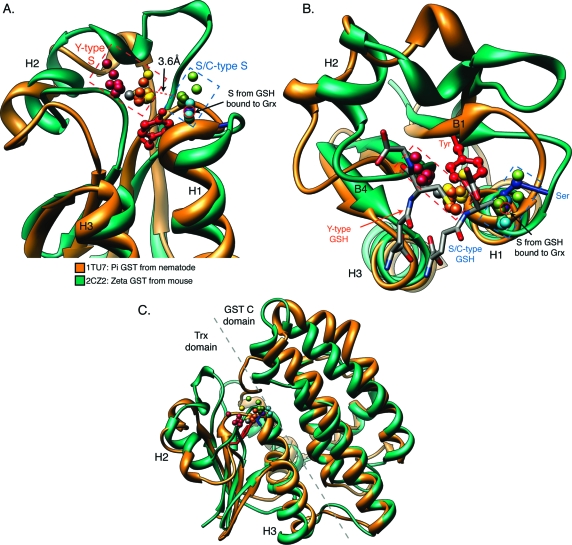
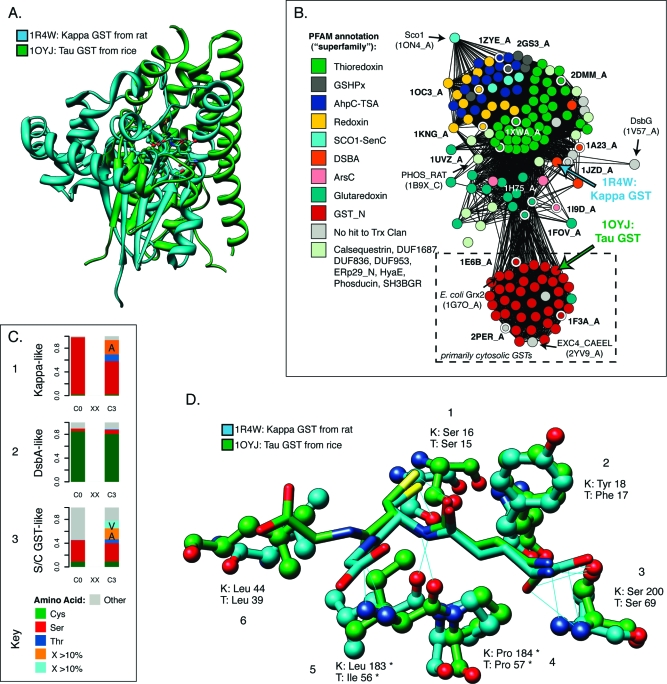
References
-
- Mannervik B.; Board P. G.; Hayes J. D.; Listowsky I.; Pearson W. R. (2005) Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 401, 1–8. - PubMed
-
- Hayes J. D.; Flanagan J. U.; Jowsey I. R. (2005) Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 45, 51–88. - PubMed
-
- Allocati N.; Federici L.; Masulli M.; Di Ilio C. (2009) Glutathione transferases in bacteria. FEBS J. 276, 58–75. - PubMed
-
- Armstrong R. N. (1991) Glutathione S-transferases: Reaction mechanism, structure, and function. Chem. Res. Toxicol. 4, 131–140. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
