

Analysis of genomic signatures in prokaryotes using multinomial regression and hierarchical clustering
- PMID: 19845945
- PMCID: PMC2770534
- DOI: 10.1186/1471-2164-10-487
Analysis of genomic signatures in prokaryotes using multinomial regression and hierarchical clustering
Abstract
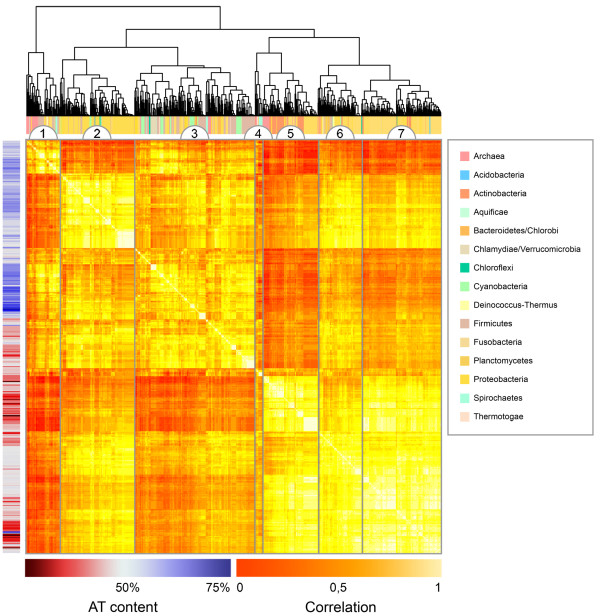
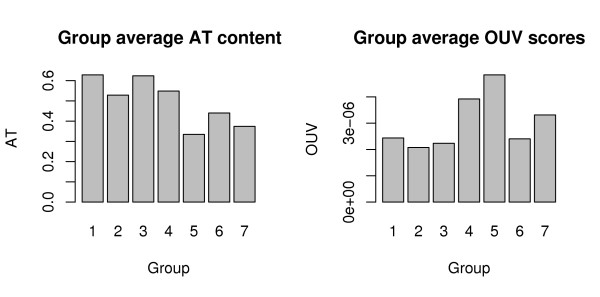
Background: Recently there has been an explosion in the availability of bacterial genomic sequences, making possible now an analysis of genomic signatures across more than 800 hundred different bacterial chromosomes, from a wide variety of environments.Using genomic signatures, we pair-wise compared 867 different genomic DNA sequences, taken from chromosomes and plasmids more than 100,000 base-pairs in length. Hierarchical clustering was performed on the outcome of the comparisons before a multinomial regression model was fitted. The regression model included the cluster groups as the response variable with AT content, phyla, growth temperature, selective pressure, habitat, sequence size, oxygen requirement and pathogenicity as predictors.
Results: Many significant factors were associated with the genomic signature, most notably AT content. Phyla was also an important factor, although considerably less so than AT content. Small improvements to the regression model, although significant, were also obtained by factors such as sequence size, habitat, growth temperature, selective pressure measured as oligonucleotide usage variance, and oxygen requirement.
Conclusion: The statistics obtained using hierarchical clustering and multinomial regression analysis indicate that the genomic signature is shaped by many factors, and this may explain the varying ability to classify prokaryotic organisms below genus level.
Figures
References
-
- DNA Databank of Japan. 2009. http://www.ddbj.nig.ac.jp/
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
