

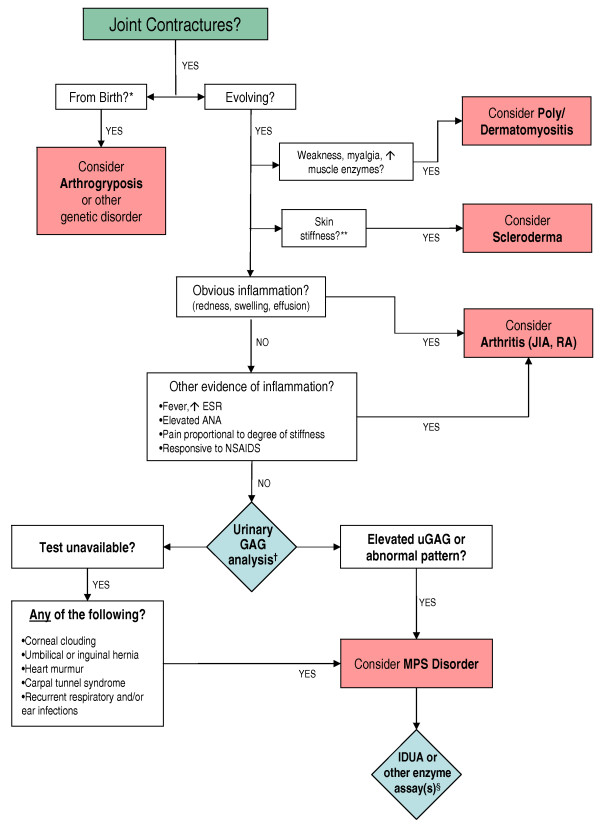
Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis
- PMID: 19852785
- PMCID: PMC2775028
- DOI: 10.1186/1546-0096-7-18
Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis
Abstract
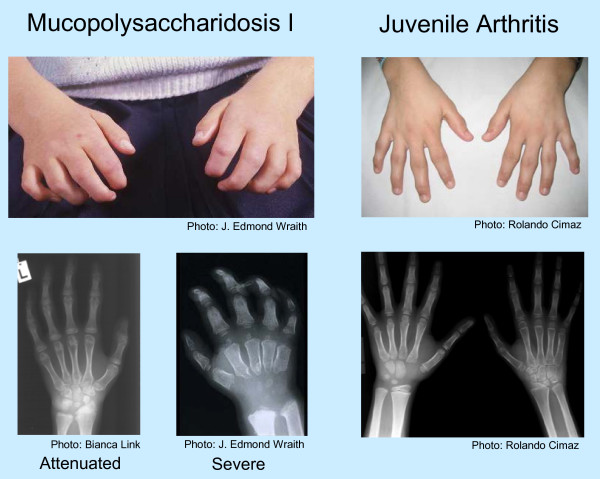
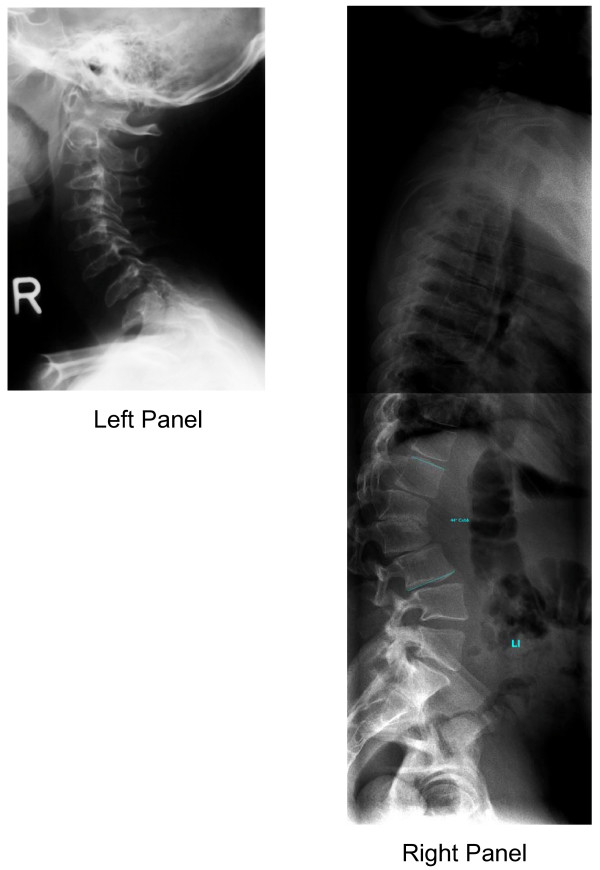
Background: Undiagnosed patients with the attenuated form of mucopolysaccharidosis (MPS) type I often have joint symptoms in childhood that prompt referral to a rheumatologist. A survey conducted by Genzyme Corporation of 60 European and Canadian rheumatologists and pediatric rheumatologists demonstrated that < 20% recognized signs and symptoms of MPS I or could identify appropriate diagnosis tests. These results prompted formation of an international working group of rheumatologists, pediatric rheumatologists, and experts on MPS I to formulate a rheumatology-based diagnostic algorithm. The resulting algorithm applies to all MPS disorders with musculoskeletal manifestations.Bone and joint manifestations are prominent among most patients with MPS disorders. These life-threatening lysosomal storage diseases are caused by deficient activity of specific enzymes involved in the degradation of glycosaminoglycans. Patients with attenuated MPS disease often experience diagnostic delays. Enzyme replacement therapy is now commercially available for MPS I (laronidase), MPS II (idursulfase), and MPS VI (galsulfase).
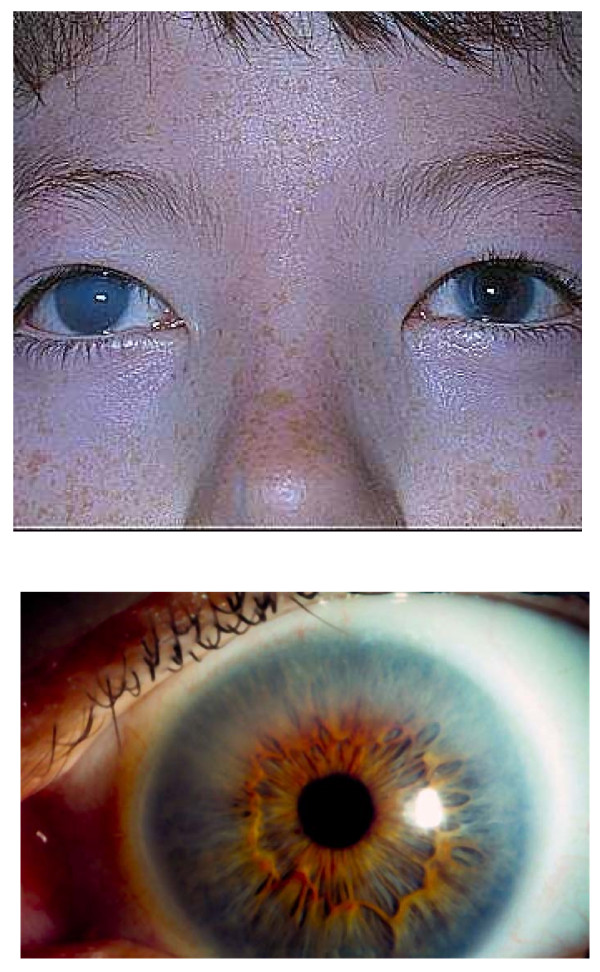
Presentation of the hypothesis: Evolving joint pain and joint contractures in the absence of inflammation should always raise the suspicion of an MPS disorder. All such patients should undergo urinary glycosaminoglycan (uGAG) analysis (not spot tests for screening) in a reputable laboratory. Elevated uGAG levels and/or an abnormal uGAG pattern confirms an MPS disorder and specific enzyme testing will determine the MPS type. If uGAG analysis is unavailable and the patient exhibits any other common sign or symptom of an MPS disorder, such as corneal clouding, history of hernia surgery, frequent respiratory and/or ear, nose and throat infections; carpal tunnel syndrome, or heart murmur, proceed directly to enzymatic testing. Refer patients with confirmed MPS to a geneticist or metabolic specialist for further evaluation and treatment.
Testing of the hypothesis: We propose that rheumatologists, pediatric rheumatologists, and orthopedists consider our diagnostic algorithm when evaluating patients with joint pain and joint contractures.
Implications of the hypothesis: Children and young adults can suffer for years and sometimes even decades with unrecognized MPS. Rheumatologists may facilitate early diagnosis of MPS based on the presenting signs and symptoms, followed by appropriate testing. Early diagnosis helps ensure prompt and appropriate treatment for these progressive and debilitating diseases.
Figures
References
-
- Cimaz R, Vijay S, Haase C, Coppa GV, Bruni S, Wraith E, Guffon N. Attenuated type I mucopolysaccharidosis in the differential diagnosis of juvenile idiopathic arthritis: a series of 13 patients with Scheie syndrome. Clin Exp Rheumatol. 2006;24:196–202. - PubMed
-
- Pastores GM, Arn P, Beck M, Clarke JT, Guffon N, Kaplan P, Muenzer J, Norato DY, Shapiro E, Thomas J, Viskochil D, Wraith JE. The MPS I registry: design, methodology, and early findings of a global disease registry for monitoring patients with mucopolysaccharidosis type I. Mol Genet Metab. 2007;91:37–47. doi: 10.1016/j.ymgme.2007.01.011. - DOI - PubMed
-
- Manger B. Rheumatological manifestations are key in the early diagnosis of mucopolysaccharidosis type I. European Musculoskeletal Review. 2008. pp. 1–6.
LinkOut - more resources
Full Text Sources
Medical
Research Materials
