

The crystal structure of the novobiocin biosynthetic enzyme NovP: the first representative structure for the TylF O-methyltransferase superfamily
- PMID: 19857499
- PMCID: PMC2813333
- DOI: 10.1016/j.jmb.2009.10.045
The crystal structure of the novobiocin biosynthetic enzyme NovP: the first representative structure for the TylF O-methyltransferase superfamily
Abstract
NovP is an S-adenosyl-l-methionine-dependent O-methyltransferase that catalyzes the penultimate step in the biosynthesis of the aminocoumarin antibiotic novobiocin. Specifically, it methylates at 4-OH of the noviose moiety, and the resultant methoxy group is important for the potency of the mature antibiotic: previous crystallographic studies have shown that this group interacts directly with the target enzyme DNA gyrase, which is a validated drug target. We have determined the high-resolution crystal structure of NovP from Streptomyces spheroides as a binary complex with its desmethylated cosubstrate S-adenosyl-l-homocysteine. The structure displays a typical class I methyltransferase fold, in addition to motifs that are consistent with a divalent-metal-dependent mechanism. This is the first representative structure of a methyltransferase from the TylF superfamily, which includes a number of enzymes implicated in the biosynthesis of antibiotics and other therapeutics. The NovP structure reveals a number of distinctive structural features that, based on sequence conservation, are likely to be characteristic of the superfamily. These include a helical 'lid' region that gates access to the cosubstrate binding pocket and an active center that contains a 3-Asp putative metal binding site. A further conserved Asp likely acts as the general base that initiates the reaction by deprotonating the 4-OH group of the noviose unit. Using in silico docking, we have generated models of the enzyme-substrate complex that are consistent with the proposed mechanism. Furthermore, these models suggest that NovP is unlikely to tolerate significant modifications at the noviose moiety, but could show increasing substrate promiscuity as a function of the distance of the modification from the methylation site. These observations could inform future attempts to utilize NovP for methylating a range of glycosylated compounds.
Copyright 2009 Elsevier Ltd. All rights reserved.
Figures

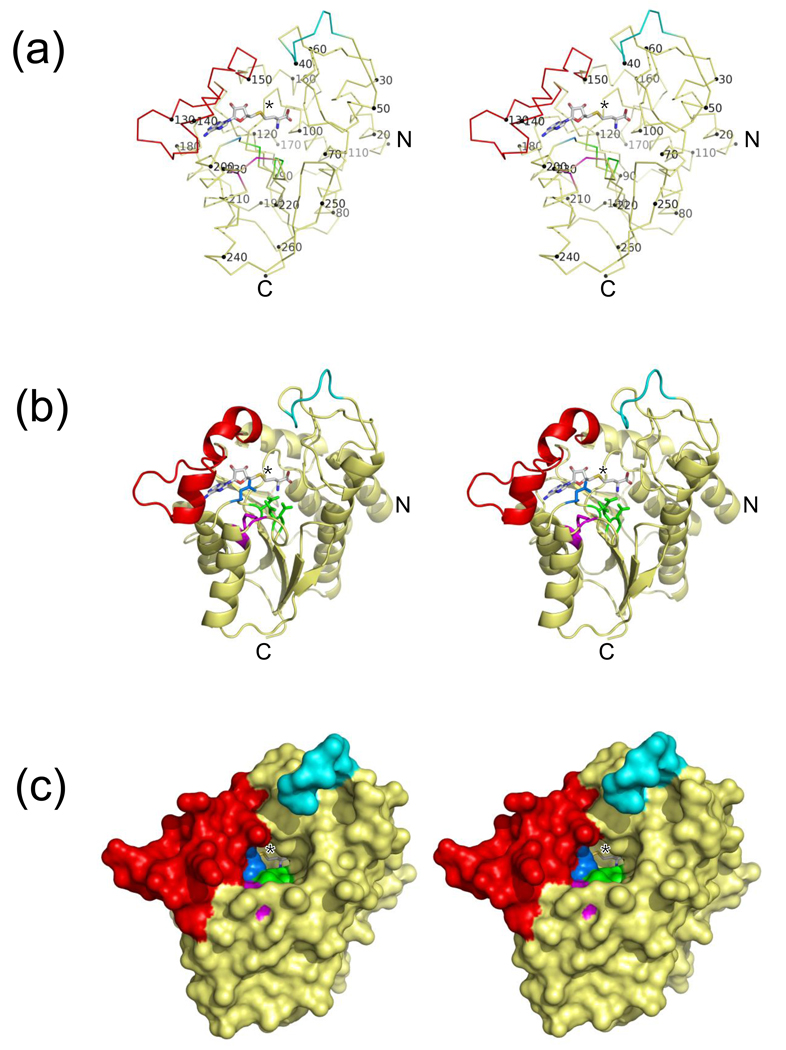



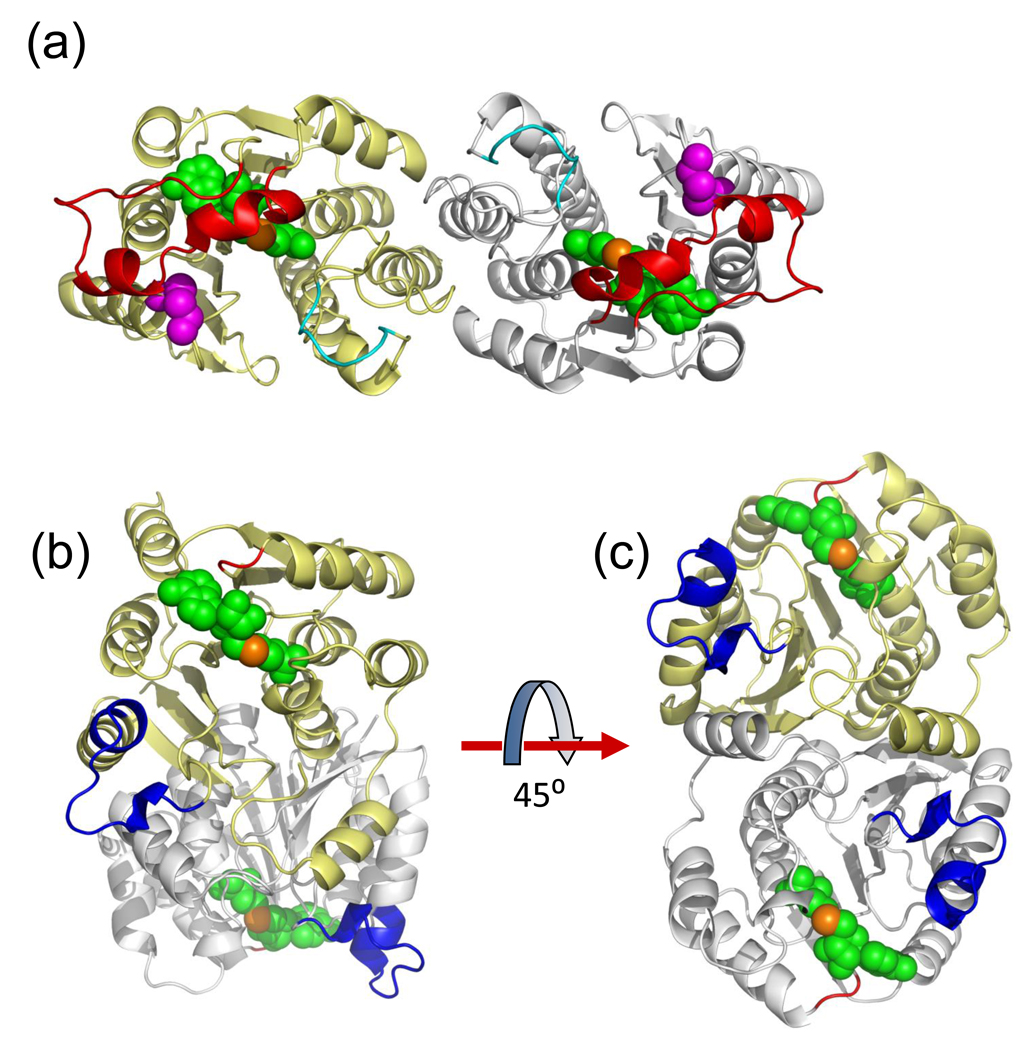




References
-
- Mannisto PT, Kaakkola S. New selective COMT inhibitors: useful adjuncts for Parkinson's disease? Trends. Pharmacol. Sci. 1989;10:54–56. - PubMed
-
- Cundliffe E, Bate N, Butler A, Fish S, Gandecha A, Merson-Davies L. The tylosin-biosynthetic genes of Streptomyces fradiae. Antonie Van Leeuwenhoek. 2001;79:229–234. - PubMed
-
- Li SM, Westrich L, Schmidt J, Kuhnt C, Heide L. Methyltransferase genes in Streptomyces rishiriensis: new coumermycin derivatives from gene-inactivation experiments. Microbiology. 2002;148:3317–3326. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
