

Genome-wide mapping of alternative splicing in Arabidopsis thaliana
- PMID: 19858364
- PMCID: PMC2798830
- DOI: 10.1101/gr.093302.109
Genome-wide mapping of alternative splicing in Arabidopsis thaliana
Abstract
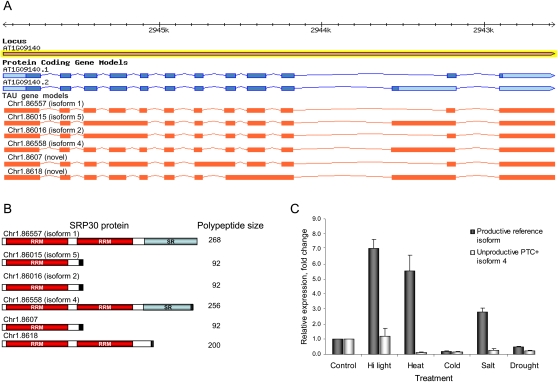
Alternative splicing can enhance transcriptome plasticity and proteome diversity. In plants, alternative splicing can be manifested at different developmental stages, and is frequently associated with specific tissue types or environmental conditions such as abiotic stress. We mapped the Arabidopsis transcriptome at single-base resolution using the Illumina platform for ultrahigh-throughput RNA sequencing (RNA-seq). Deep transcriptome sequencing confirmed a majority of annotated introns and identified thousands of novel alternatively spliced mRNA isoforms. Our analysis suggests that at least approximately 42% of intron-containing genes in Arabidopsis are alternatively spliced; this is significantly higher than previous estimates based on cDNA/expressed sequence tag sequencing. Random validation confirmed that novel splice isoforms empirically predicted by RNA-seq can be detected in vivo. Novel introns detected by RNA-seq were substantially enriched in nonconsensus terminal dinucleotide splice signals. Alternative isoforms with premature termination codons (PTCs) comprised the majority of alternatively spliced transcripts. Using an example of an essential circadian clock gene, we show that intron retention can generate relatively abundant PTC(+) isoforms and that this specific event is highly conserved among diverse plant species. Alternatively spliced PTC(+) isoforms can be potentially targeted for degradation by the nonsense mediated mRNA decay (NMD) surveillance machinery or regulate the level of functional transcripts by the mechanism of regulated unproductive splicing and translation (RUST). We demonstrate that the relative ratios of the PTC(+) and reference isoforms for several key regulatory genes can be considerably shifted under abiotic stress treatments. Taken together, our results suggest that like in animals, NMD and RUST may be widespread in plants and may play important roles in regulating gene expression.
Figures






References
-
- Alexandrov NN, Troukhan ME, Brover VV, Tatarinova T, Flavell RB, Feldmann KA. Features of Arabidopsis genes and genome discovered using full-length cDNAs. Plant Mol Biol. 2006;60:69–85. - PubMed
-
- Ali GS, Reddy ASN. ATP, phosphorylation and transcription regulate the mobility of plant splicing factors. J Cell Sci. 2006;119:3527–3538. - PubMed
-
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources