

The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance
- PMID: 19861693
- PMCID: PMC2852205
- DOI: 10.1210/er.2009-0003
The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance
Abstract
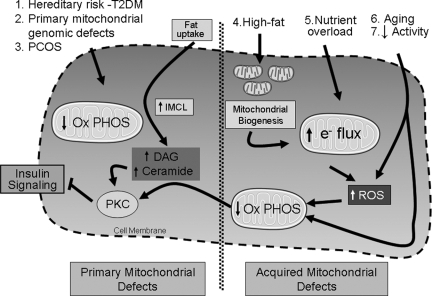
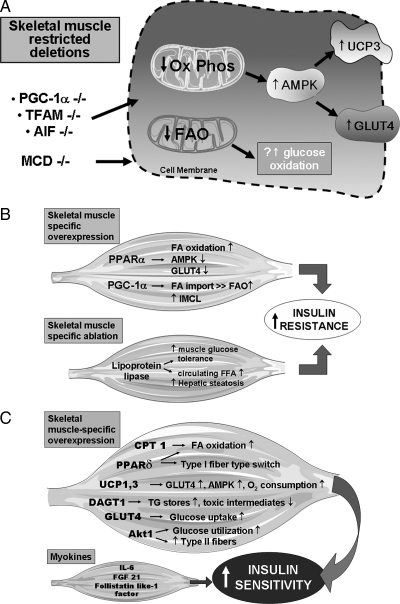
Multiple organs contribute to the development of peripheral insulin resistance, with the major contributors being skeletal muscle, liver, and adipose tissue. Because insulin resistance usually precedes the development of type 2 diabetes mellitus (T2DM) by many years, understanding the pathophysiology of insulin resistance should enable development of therapeutic strategies to prevent disease progression. Some subjects with mitochondrial genomic variants/defects and a subset of lean individuals with hereditary predisposition to T2DM exhibit skeletal muscle mitochondrial dysfunction early in the course of insulin resistance. In contrast, in the majority of subjects with T2DM the plurality of evidence implicates skeletal muscle mitochondrial dysfunction as a consequence of perturbations associated with T2DM, and these mitochondrial deficits then contribute to subsequent disease progression. We review the affirmative and contrarian data regarding skeletal muscle mitochondrial biology in the pathogenesis of insulin resistance and explore potential therapeutic options to intrinsically modulate mitochondria as a strategy to combat insulin resistance. Furthermore, an overview of restricted molecular manipulations of skeletal muscle metabolic and mitochondrial biology offers insight into the mitochondrial role in metabolic substrate partitioning and in promoting innate adaptive and maladaptive responses that collectively regulate peripheral insulin sensitivity. We conclude that skeletal muscle mitochondrial dysfunction is not generally a major initiator of the pathophysiology of insulin resistance, although its dysfunction is integral to this pathophysiology and it remains an intriguing target to reverse/delay the progressive perturbations synonymous with T2DM.
Figures
References
-
- Reaven GM 1995 Pathophysiology of insulin resistance in human disease. Physiol Rev 75:473–486 - PubMed
-
- Fernández-Real JM, Pickup JC 2008 Innate immunity, insulin resistance and type 2 diabetes. Trends Endocrinol Metab 19:10–16 - PubMed
-
- Karlsson HK, Ahlsén M, Zierath JR, Wallberg-Henriksson H, Koistinen HA 2006 Insulin signaling and glucose transport in skeletal muscle from first-degree relatives of type 2 diabetic patients. Diabetes 55:1283–1288 - PubMed
-
- Meyer C, Dostou JM, Welle SL, Gerich JE 2002 Role of human liver, kidney, and skeletal muscle in postprandial glucose homeostasis. Am J Physiol Endocrinol Metab 282:E419–E427 - PubMed
-
- Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG 1990 Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med 322:223–228 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
