

Beta-catenin up-regulates Atoh1 expression in neural progenitor cells by interaction with an Atoh1 3' enhancer
- PMID: 19864427
- PMCID: PMC2804186
- DOI: 10.1074/jbc.M109.059055
Beta-catenin up-regulates Atoh1 expression in neural progenitor cells by interaction with an Atoh1 3' enhancer
Abstract
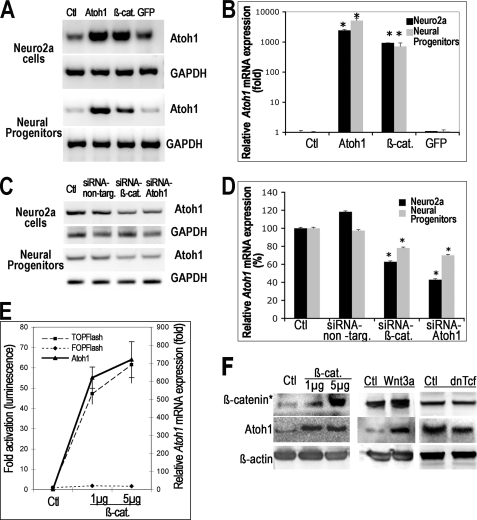
Atoh1, a basic helix-loop-helix transcription factor, plays a critical role in the differentiation of several epithelial and neural cell types. We found that beta-catenin, the key mediator of the canonical Wnt pathway, increased expression of Atoh1 in mouse neuroblastoma cells and neural progenitor cells, and baseline Atoh1 expression was decreased by siRNA directed at beta-catenin. The up-regulation of Atoh1 was caused by an interaction of beta-catenin with the Atoh1 enhancer that could be demonstrated by chromatin immunoprecipitation. We found that two putative Tcf-Lef sites in the 3' enhancer of the Atoh1 gene displayed an affinity for beta-catenin and were critical for the activation of Atoh1 transcription because mutation of either site decreased expression of a reporter gene downstream of the enhancer. Tcf-Lef co-activators were found in the complex that bound to these sites in the DNA together with beta-catenin. Inhibition of Notch signaling, which has previously been shown to induce bHLH transcription factor expression, increased beta-catenin expression in progenitor cells of the nervous system. Because this could be a mechanism for up-regulation of Atoh1 after inhibition of Notch, we tested whether siRNA to beta-catenin prevented the increase in Atoh1 and found that beta-catenin expression was required for increased expression of Atoh1 after Notch inhibition.
Figures





References
-
- Bermingham N. A., Hassan B. A., Price S. D., Vollrath M. A., Ben-Arie N., Eatock R. A., Bellen H. J., Lysakowski A., Zoghbi H. Y. (1999) Science 284, 1837–1841 - PubMed
-
- Yang Q., Bermingham N. A., Finegold M. J., Zoghbi H. Y. (2001) Science 294, 2155–2158 - PubMed
-
- Bertrand N., Castro D. S., Guillemot F. (2002) Nat. Rev. Neurosci. 3, 517–530 - PubMed
-
- Ross S. E., Greenberg M. E., Stiles C. D. (2003) Neuron 39, 13–25 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
