

Exploitation of binding energy for catalysis and design
- PMID: 19865174
- PMCID: PMC2771326
- DOI: 10.1038/nature08508
Exploitation of binding energy for catalysis and design
Abstract
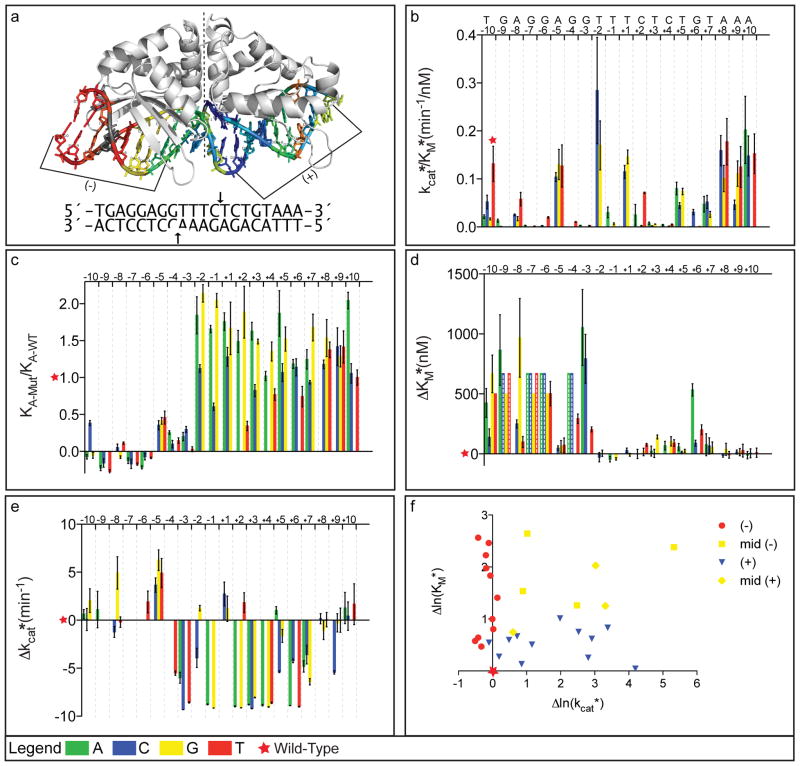
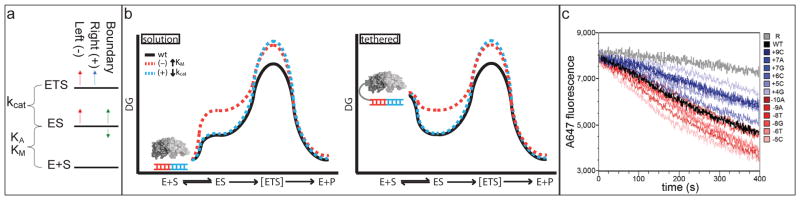
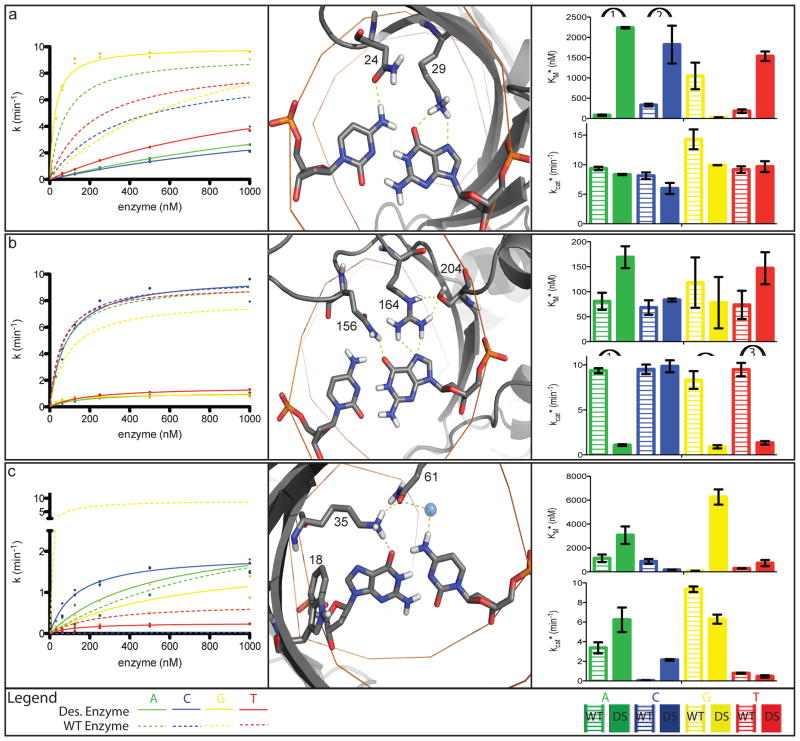
Enzymes use substrate-binding energy both to promote ground-state association and to stabilize the reaction transition state selectively. The monomeric homing endonuclease I-AniI cleaves with high sequence specificity in the centre of a 20-base-pair (bp) DNA target site, with the amino (N)-terminal domain of the enzyme making extensive binding interactions with the left (-) side of the target site and the similarly structured carboxy (C)-terminal domain interacting with the right (+) side. Here we show that, despite the approximate twofold symmetry of the enzyme-DNA complex, there is almost complete segregation of interactions responsible for substrate binding to the (-) side of the interface and interactions responsible for transition-state stabilization to the (+) side. Although single base-pair substitutions throughout the entire DNA target site reduce catalytic efficiency, mutations in the (-) DNA half-site almost exclusively increase the dissociation constant (K(D)) and the Michaelis constant under single-turnover conditions (K(M)*), and those in the (+) half-site primarily decrease the turnover number (k(cat)*). The reduction of activity produced by mutations on the (-) side, but not mutations on the (+) side, can be suppressed by tethering the substrate to the endonuclease displayed on the surface of yeast. This dramatic asymmetry in the use of enzyme-substrate binding energy for catalysis has direct relevance to the redesign of endonucleases to cleave genomic target sites for gene therapy and other applications. Computationally redesigned enzymes that achieve new specificities on the (-) side do so by modulating K(M)*, whereas redesigns with altered specificities on the (+) side modulate k(cat)*. Our results illustrate how classical enzymology and modern protein design can each inform the other.
Figures
References
-
- Jencks WP. Mechanism of enzyme action. Annu Rev Biochem. 1963;32:639–676. - PubMed
-
- Wells TN, Fersht AR. Use of binding energy in catalysis measured by mutagenesis of tyrosyl-tRNA synthetase. Biochemistry. 1986;25:1881–1886. - PubMed
-
- Fersht AR. Relationships between apparent binding energies measured in site-directed mutagenesis experiments and energetics of binding and catalysis. Biochemistry. 1988;27:1577–1580. - PubMed
-
- Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science. 2003;301:1196–1202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
