

CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel-induced cytotoxicity
- PMID: 19866475
- PMCID: PMC2931415
- DOI: 10.1002/pros.21077
CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel-induced cytotoxicity
Abstract
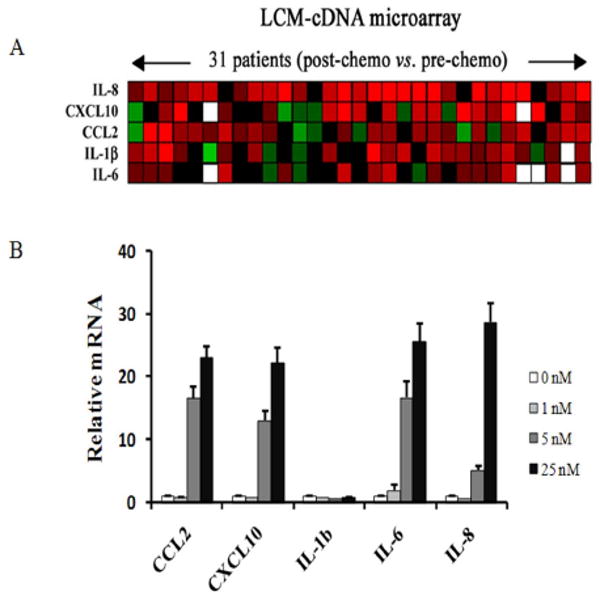
Background: Metastatic prostate cancer is either inherently resistant to chemotherapy or rapidly acquires this phenotype after chemotherapy exposure. In this study, we identified a docetaxel-induced resistance mechanism centered on CCL2.
Methods: We compared the gene expression profiles in individual human prostate cancer specimens before and after exposure to chemotherapy collected from previously untreated patients who participated in a clinical trial of preoperative chemotherapy. Subsequently, we used the gain- and loss-of-function approach in vitro to identify a potential mechanism underlying chemotherapy resistance.
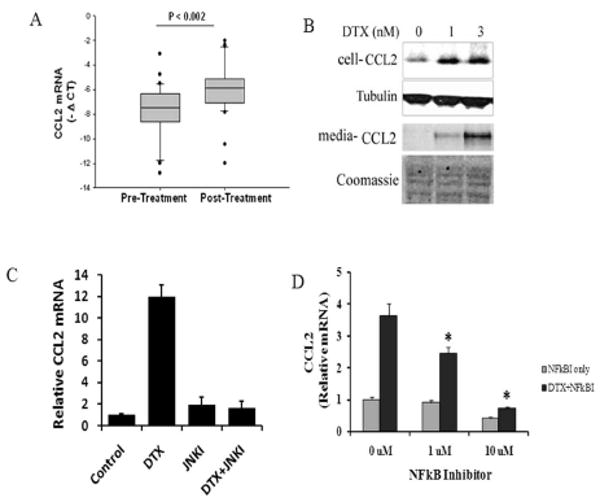
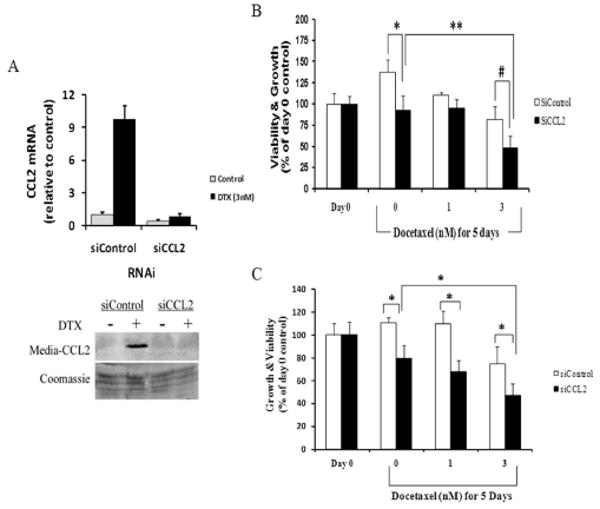
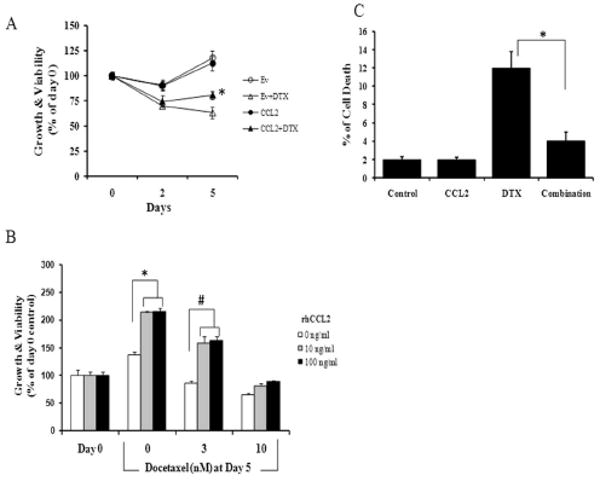
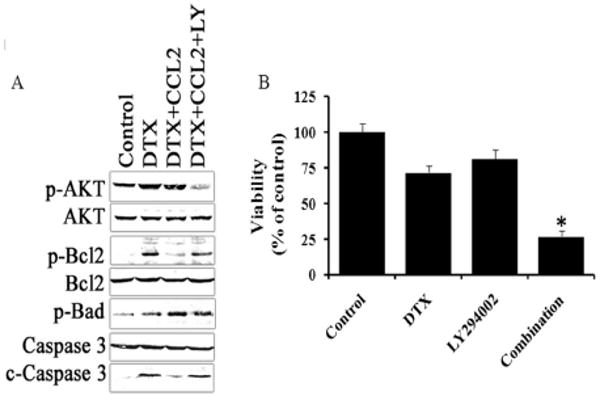
Results: Among the molecular signatures associated with treatment, several genes that regulate the inflammatory response and chemokine activity were upregulated including a significant increase in transcripts encoding the CC chemokine CCL2. Docetaxel increased CCL2 expression in prostate cancer cell lines in vitro. CCL2-specific siRNA inhibited LNCaP and LAPC4 cell proliferation and enhanced the growth inhibitory effect of low-dose docetaxel. In contrast, overexpression of CCL2 or recombinant CCL2 protein stimulated prostate cancer cell proliferation and rescued cells from docetaxel-induced cytotoxicity. This protective effect of CCL2 was associated with activation of the ERK/MAP kinase and PI3K/AKT, inhibition of docetaxel-induced Bcl2 phosphorylation at serine 70, phosphorylation of Bad, and activation of caspase-3. The addition of a PI3K/AKT inhibitor Ly294002 reversed the CCL2 protection and was additive to docetaxel-induced toxicity.
Conclusion: These results support a mechanism of chemotherapy resistance mediated by cellular stress responses involving the induction of CCL2 expression and suggest that inhibiting CCL2 activity could enhance therapeutic responses to taxane-based therapy.
Prostate 70: 433-442, 2010. (c) 2009 Wiley-Liss, Inc.
Figures

Similar articles
-
Targeting the Vav3 oncogene enhances docetaxel-induced apoptosis through the inhibition of androgen receptor phosphorylation in LNCaP prostate cancer cells under chronic hypoxia.Mol Cancer. 2013 Apr 8;12:27. doi: 10.1186/1476-4598-12-27. Mol Cancer. 2013. PMID: 23566222 Free PMC article.
-
ID1 enhances docetaxel cytotoxicity in prostate cancer cells through inhibition of p21.Cancer Res. 2010 Apr 15;70(8):3239-48. doi: 10.1158/0008-5472.CAN-09-3186. Epub 2010 Apr 13. Cancer Res. 2010. PMID: 20388787 Free PMC article.
-
CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells.Cancer Sci. 2019 Jan;110(1):279-288. doi: 10.1111/cas.13876. Epub 2018 Dec 7. Cancer Sci. 2019. PMID: 30426599 Free PMC article.
-
Gene expression profiling of the androgen independent prostate cancer cells demonstrates complex mechanisms mediating resistance to docetaxel.Cancer Biol Ther. 2011 Jan 15;11(2):204-12. doi: 10.4161/cbt.11.2.13750. Epub 2011 Jan 15. Cancer Biol Ther. 2011. PMID: 21057205 Free PMC article.
-
Second-line chemotherapy in metastatic docetaxel-resistant prostate cancer: a review.Med Oncol. 2012 Jun;29(2):776-85. doi: 10.1007/s12032-011-9855-6. Epub 2011 Feb 20. Med Oncol. 2012. PMID: 21336988 Review.
Cited by
-
Inhibition of CCL2 signaling in combination with docetaxel treatment has profound inhibitory effects on prostate cancer growth in bone.Int J Mol Sci. 2013 May 21;14(5):10483-96. doi: 10.3390/ijms140510483. Int J Mol Sci. 2013. PMID: 23698775 Free PMC article.
-
Immunotherapy by mesenchymal stromal cell delivery of oncolytic viruses for treating metastatic tumors.Mol Ther Oncolytics. 2022 Mar 19;25:78-97. doi: 10.1016/j.omto.2022.03.008. eCollection 2022 Jun 16. Mol Ther Oncolytics. 2022. PMID: 35434272 Free PMC article. Review.
-
Priming cancer cells for drug resistance: role of the fibroblast niche.Front Biol (Beijing). 2014 Feb 1;9(2):114-126. doi: 10.1007/s11515-014-1300-8. Front Biol (Beijing). 2014. PMID: 25045348 Free PMC article.
-
The multifaceted roles of the chemokines CCL2 and CXCL12 in osteophilic metastatic cancers.Cancer Metastasis Rev. 2021 Jun;40(2):427-445. doi: 10.1007/s10555-021-09974-2. Epub 2021 May 11. Cancer Metastasis Rev. 2021. PMID: 33973098 Review.
-
Neutralizing tumor-promoting chronic inflammation: a magic bullet?Science. 2013 Jan 18;339(6117):286-91. doi: 10.1126/science.1232227. Science. 2013. PMID: 23329041 Free PMC article. Review.
References
-
- Mackler NJ, Pienta KJ. Drug insight: Use of docetaxel in prostate and urothelial cancers. Nat Clin Pract Urol. 2005;2(2):92–100. quiz 101 p following 112. - PubMed
-
- Pienta KJ, Smith DC. Advances in prostate cancer chemotherapy: a new era begins. CA Cancer J Clin. 2005;55(5):300–318. quiz 323-305. - PubMed
-
- Beer TM, El-Geneidi M, Eilers KM. Docetaxel (taxotere) in the treatment of prostate cancer. Expert Rev Anticancer Ther. 2003;3(3):261–268. - PubMed
-
- Garzotto M, Myrthue A, Higano CS, Beer TM. Neoadjuvant mitoxantrone and docetaxel for high-risk localized prostate cancer. Urol Oncol. 2006;24(3):254–259. - PubMed
-
- Huang CY, Beer TM, Higano CS, True LD, Vessella R, Lange PH, Garzotto M, Nelson PS. Molecular alterations in prostate carcinomas that associate with in vivo exposure to chemotherapy: identification of a cytoprotective mechanism involving growth differentiation factor 15. Clin Cancer Res. 2007;13(19):5825–5833. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
