

Milder clinical hyperimmunoglobulin E syndrome phenotype is associated with partial interleukin-17 deficiency
- PMID: 19878510
- PMCID: PMC2802694
- DOI: 10.1111/j.1365-2249.2009.04043.x
Milder clinical hyperimmunoglobulin E syndrome phenotype is associated with partial interleukin-17 deficiency
Erratum in
- Clin Exp Immunol. 2011 May;164(2):289. Marijnissen, R [corrected to Marijnissen R J]
Abstract
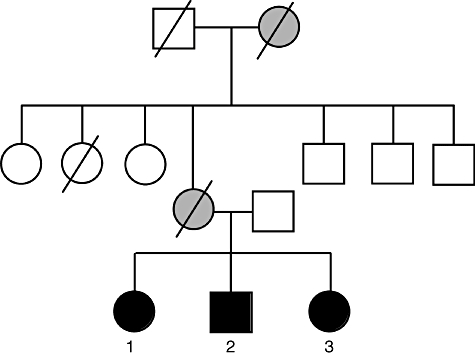
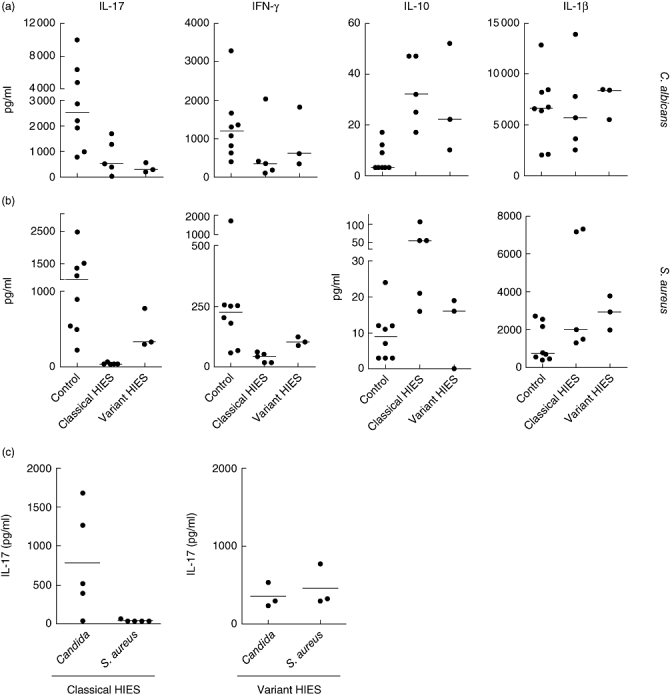
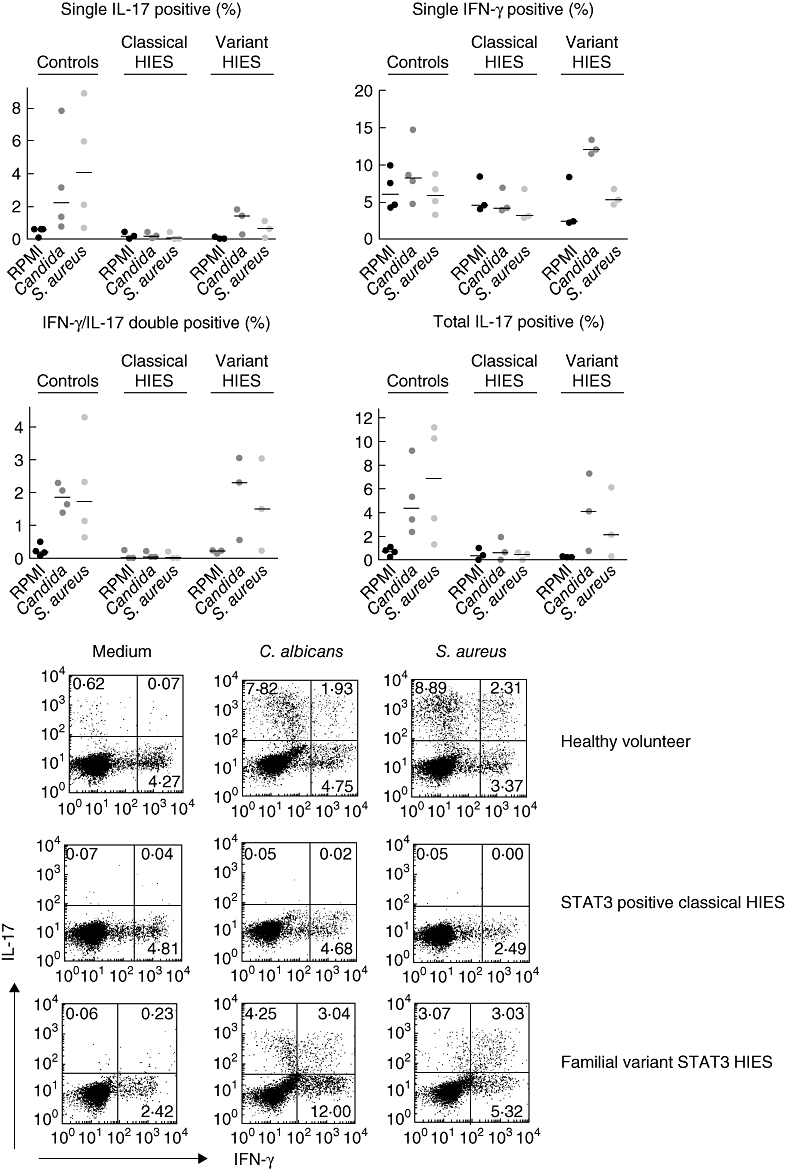
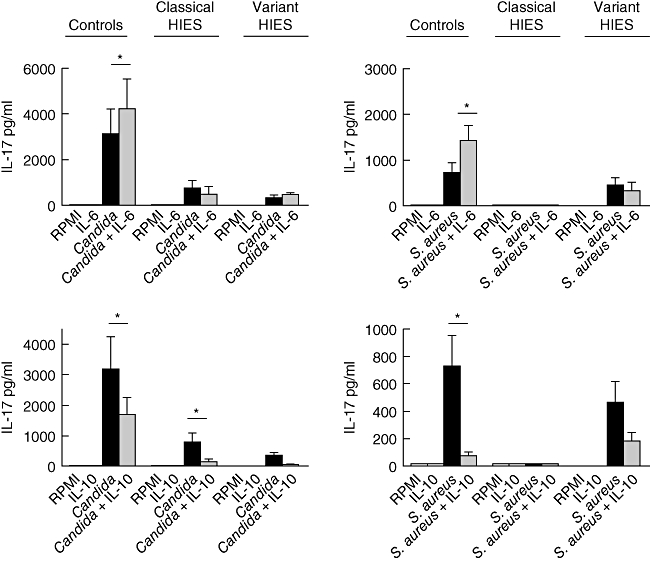
Mutations in the signal transducer and activator of transcription 3 (STAT3) were reported to cause hyperimmunoglobulin E syndrome (HIES). The present study investigates T helper type 17 (Th17) responses triggered by the relevant stimuli Staphylococcus aureus and Candidia albicans in five 'classical' HIES patients, and a family with three patients who all had a milder HIES phenotype. We demonstrate that patients with various forms of HIES have different defects in their Th17 response to S. aureus and C. albicans, and this is in line with the clinical features of the disease. Interestingly, a partial deficiency of interleukin (IL)-17 production, even when associated with STAT3 mutations, leads to a milder clinical phenotype. We also observed defective Th17 responses in patients with the 'classical' presentation of the disease but without STAT3 mutations. These data demonstrate that defective IL-17 production in response to specific pathogens can differ between patients with HIES and that the extent of the defective Th17 response determines their clinical phenotype.
Figures
References
-
- Davis SD, Schaller J, Wedgwood RJ. Job's syndrome: recurrent ‘cold’ staphylococcal abcesses. Lancet. 1966;1:1013–15. - PubMed
-
- Buckley RH, Wray BB, Belmaker EZ. Extreme hyperimmunoglobulinemia E and undue susceptibility to infection. Pediatrics. 1972;49:59–70. - PubMed
-
- Grimbacher B, Holland SM, Gallin JI, et al. Hyper-IgE syndrome with recurrent infections – an autosomal dominant multisystem disorder. N Engl J Med. 1999;340:692–702. - PubMed
-
- Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–62. - PubMed
-
- Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med. 2007;357:1608–19. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
