

T cell-microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening?
- PMID: 19879804
- PMCID: PMC4126423
- DOI: 10.1016/j.it.2009.09.003
T cell-microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening?
Abstract
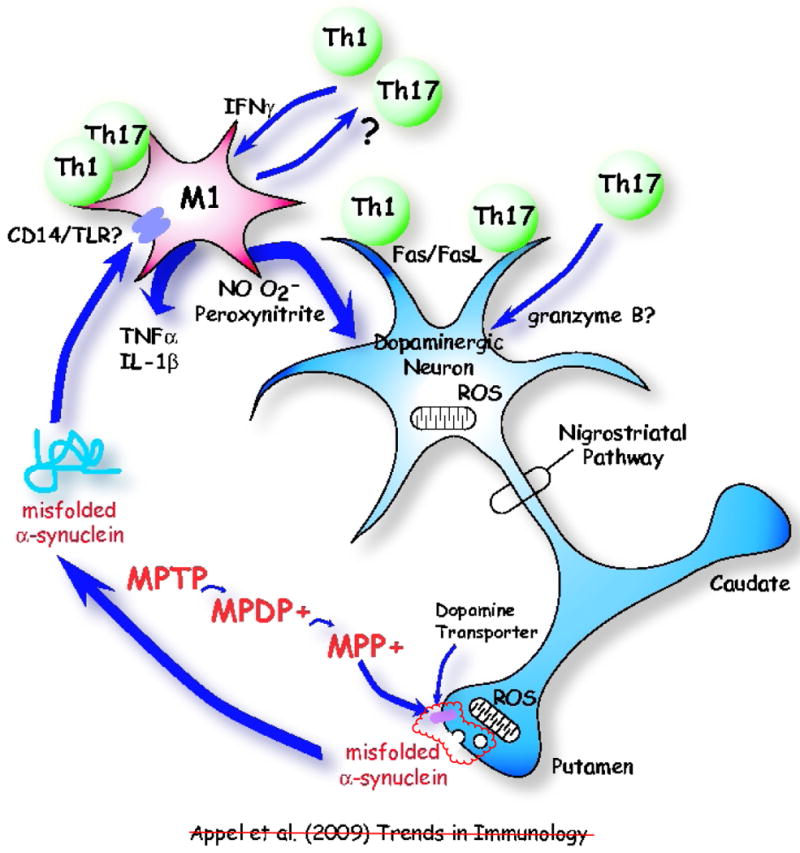
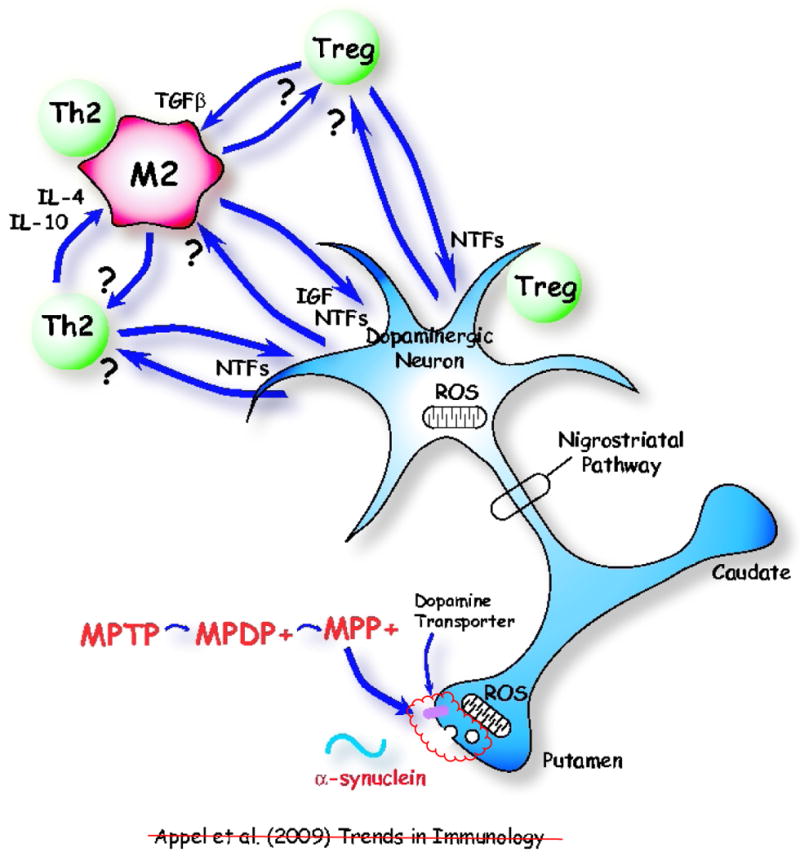
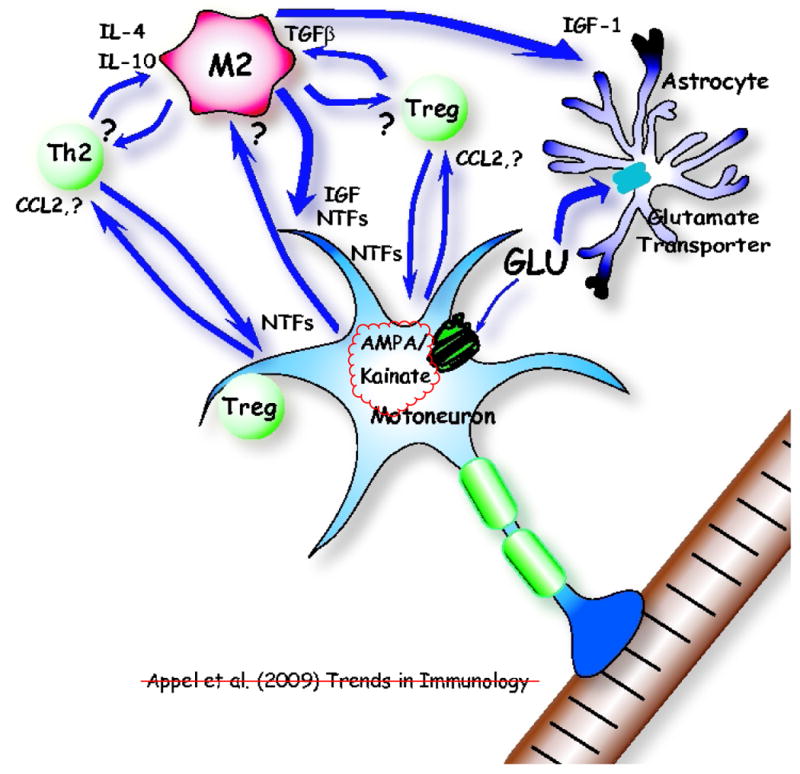
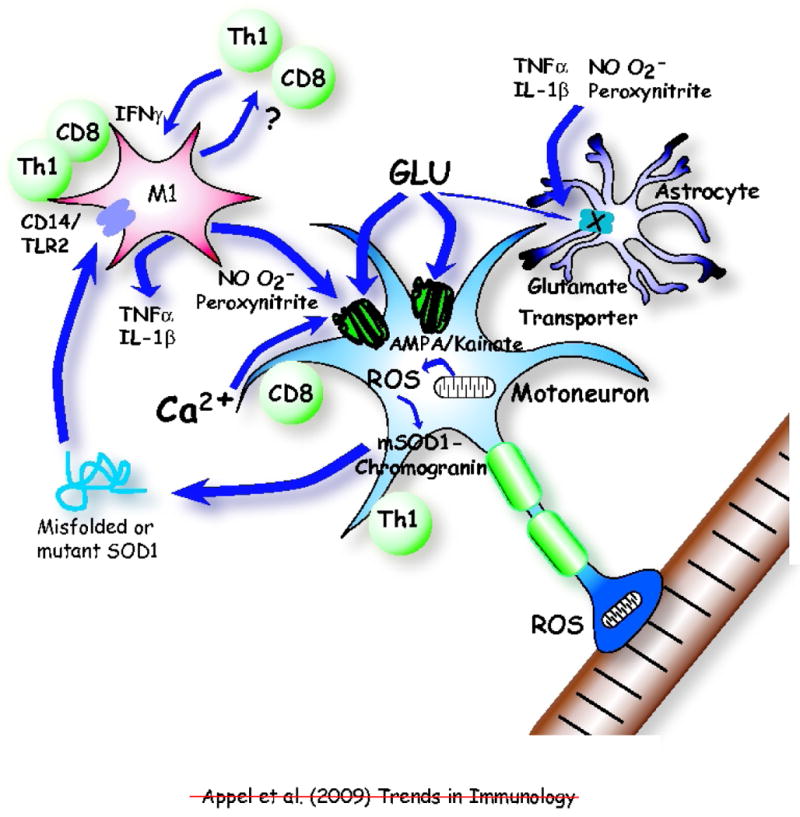
Neuroinflammation is a pathological hallmark in Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS), and is characterized by activated microglia and infiltrating T cells at sites of neuronal injury. In PD and ALS, neurons do not die alone; neuronal injury is non-cell-autonomous and depends on a well-orchestrated dialogue in which neuronally secreted misfolded proteins activate microglia and initiate a self-propagating cycle of neurotoxicity. Diverse populations and phenotypes of CD4(+) T cells crosstalk with microglia, and depending on their activation status, influence this dialogue and promote neuroprotection or neurotoxicity. A greater understanding of the T cell population that mediates these effects, as well as the molecular signals involved should provide targets for neuroprotective immunomodulation to treat these devastating neurodegenerative diseases.
Figures
References
-
- Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. - PubMed
-
- Boillée S, et al. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. - PubMed
-
- Langston JW, et al. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. - PubMed
-
- Dauer W, Przedborski S. Parkinson's disease: Mechanisms and Models. Neuron. 2003;39:889–909. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
