

fPOP: footprinting functional pockets of proteins by comparative spatial patterns
- PMID: 19880384
- PMCID: PMC2808891
- DOI: 10.1093/nar/gkp900
fPOP: footprinting functional pockets of proteins by comparative spatial patterns
Abstract
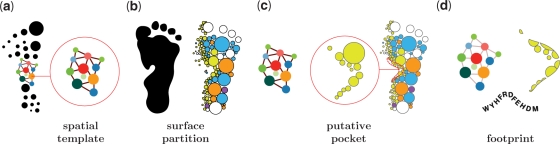
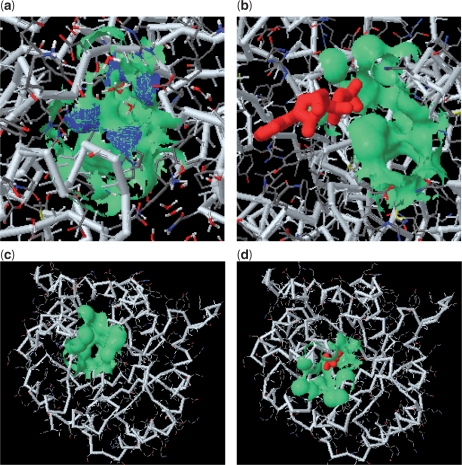
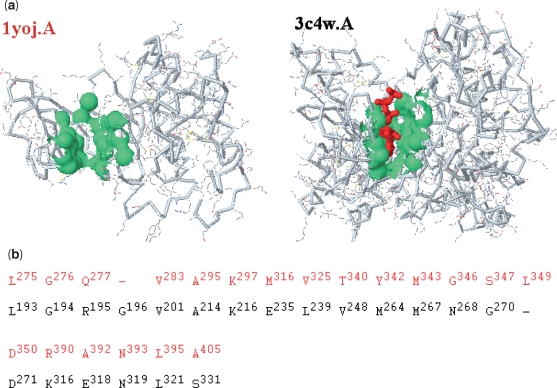
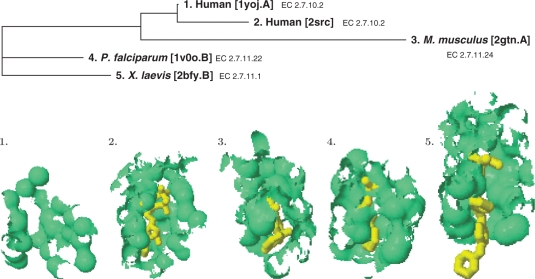
fPOP (footprinting Pockets Of Proteins, http://pocket.uchicago.edu/fpop/) is a relational database of the protein functional surfaces identified by analyzing the shapes of binding sites in approximately 42,700 structures, including both holo and apo forms. We previously used a purely geometric method to extract the spatial patterns of functional surfaces (split pockets) in approximately 19,000 bound structures and constructed a database, SplitPocket (http://pocket.uchicago.edu/). These functional surfaces are now used as spatial templates to predict the binding surfaces of unbound structures. To conduct a shape comparison, we use the Smith-Waterman algorithm to footprint an unbound pocket fragment with those of the functional surfaces in SplitPocket. The pairwise alignment of the unbound and bound pocket fragments is used to evaluate the local structural similarity via geometric matching. The final results of our large-scale computation, including approximately 90,000 identified or predicted functional surfaces, are stored in fPOP. This database provides an easily accessible resource for studying functional surfaces, assessing conformational changes between bound and unbound forms and analyzing functional divergence. Moreover, it may facilitate the exploration of the physicochemical textures of molecules and the inference of protein function. Finally, our approach provides a framework for classification of proteins into families on the basis of their functional surfaces.
Figures
Similar articles
-
SplitPocket: identification of protein functional surfaces and characterization of their spatial patterns.Nucleic Acids Res. 2009 Jul;37(Web Server issue):W384-9. doi: 10.1093/nar/gkp308. Epub 2009 Apr 30. Nucleic Acids Res. 2009. PMID: 19406922 Free PMC article.
-
Identification of protein functional surfaces by the concept of a split pocket.Proteins. 2009 Sep;76(4):959-76. doi: 10.1002/prot.22402. Proteins. 2009. PMID: 19326458
-
ComSin: database of protein structures in bound (complex) and unbound (single) states in relation to their intrinsic disorder.Nucleic Acids Res. 2010 Jan;38(Database issue):D283-7. doi: 10.1093/nar/gkp963. Epub 2009 Nov 11. Nucleic Acids Res. 2010. PMID: 19906708 Free PMC article.
-
Chapter 4. Predicting and characterizing protein functions through matching geometric and evolutionary patterns of binding surfaces.Adv Protein Chem Struct Biol. 2008;75:107-41. doi: 10.1016/S0065-3233(07)75004-0. Epub 2009 Feb 26. Adv Protein Chem Struct Biol. 2008. PMID: 20731991 Free PMC article. Review.
-
Mass Spectrometry-Based Fast Photochemical Oxidation of Proteins (FPOP) for Higher Order Structure Characterization.Acc Chem Res. 2018 Mar 20;51(3):736-744. doi: 10.1021/acs.accounts.7b00593. Epub 2018 Feb 16. Acc Chem Res. 2018. PMID: 29450991 Free PMC article. Review.
Cited by
-
CavitySpace: A Database of Potential Ligand Binding Sites in the Human Proteome.Biomolecules. 2022 Jul 11;12(7):967. doi: 10.3390/biom12070967. Biomolecules. 2022. PMID: 35883523 Free PMC article.
-
Normal Modes Expose Active Sites in Enzymes.PLoS Comput Biol. 2016 Dec 21;12(12):e1005293. doi: 10.1371/journal.pcbi.1005293. eCollection 2016 Dec. PLoS Comput Biol. 2016. PMID: 28002427 Free PMC article.
-
fpocket: online tools for protein ensemble pocket detection and tracking.Nucleic Acids Res. 2010 Jul;38(Web Server issue):W582-9. doi: 10.1093/nar/gkq383. Epub 2010 May 16. Nucleic Acids Res. 2010. PMID: 20478829 Free PMC article.
-
VirtuousPocketome: a computational tool for screening protein-ligand complexes to identify similar binding sites.Sci Rep. 2024 Mar 15;14(1):6296. doi: 10.1038/s41598-024-56893-7. Sci Rep. 2024. PMID: 38491261 Free PMC article.
-
Synchrotron X-ray footprinting as a method to visualize water in proteins.J Synchrotron Radiat. 2016 Sep 1;23(Pt 5):1056-69. doi: 10.1107/S1600577516009024. Epub 2016 Jul 27. J Synchrotron Radiat. 2016. PMID: 27577756 Free PMC article.
References
-
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. - PubMed
