

Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy
- PMID: 19881493
- PMCID: PMC3290906
- DOI: 10.1038/nm.2052
Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy
Abstract
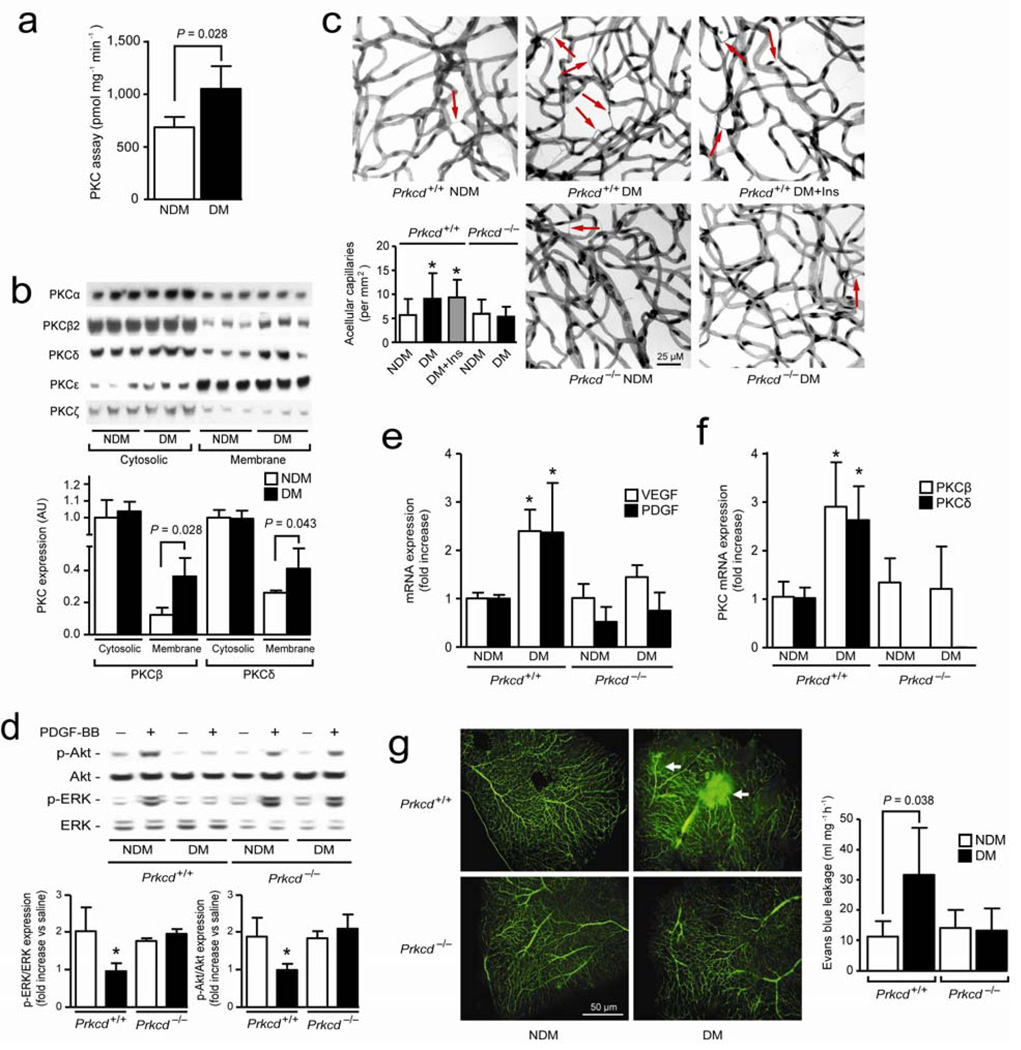
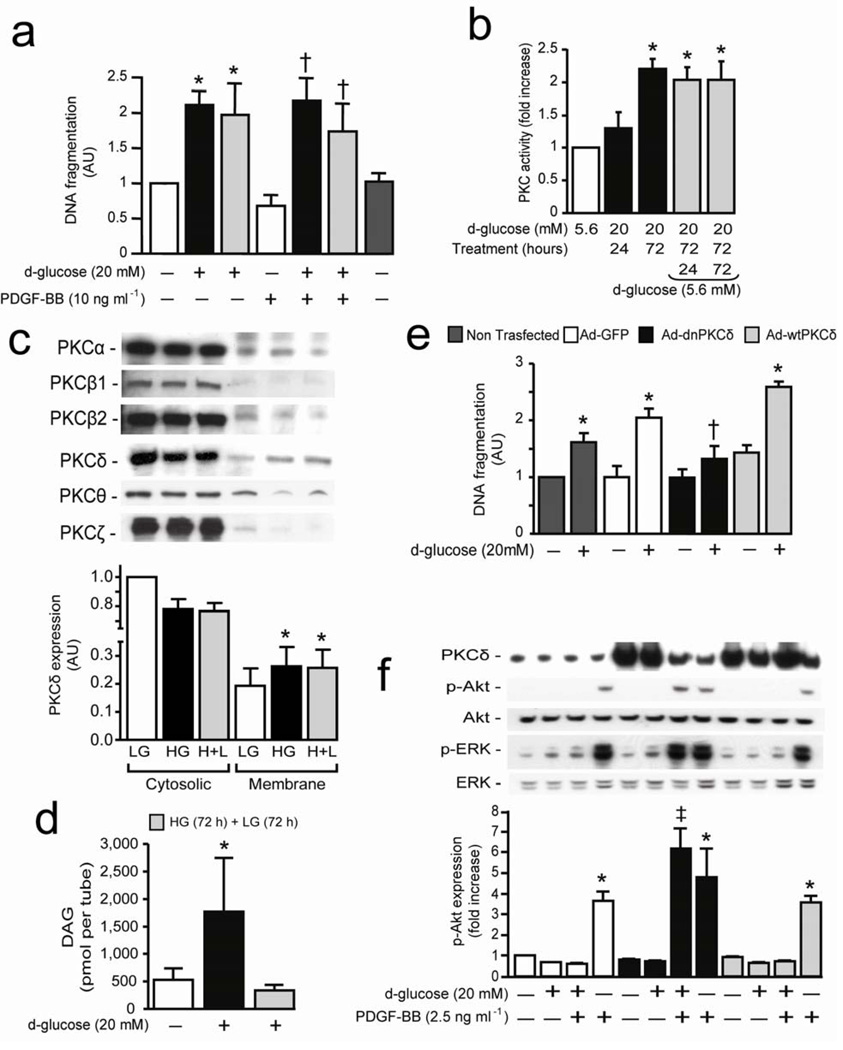
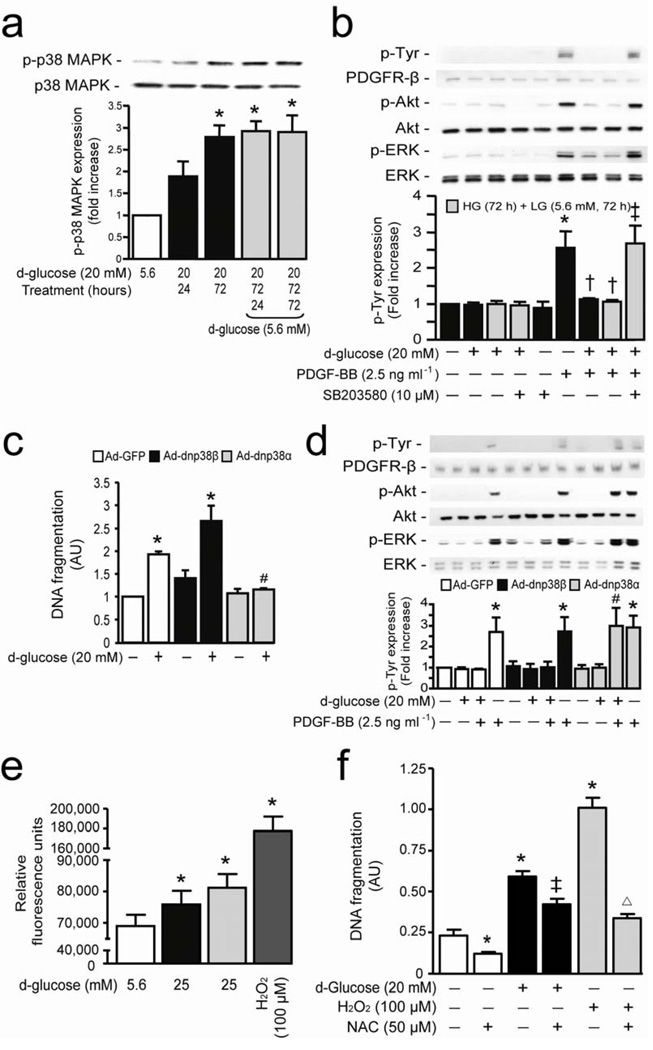
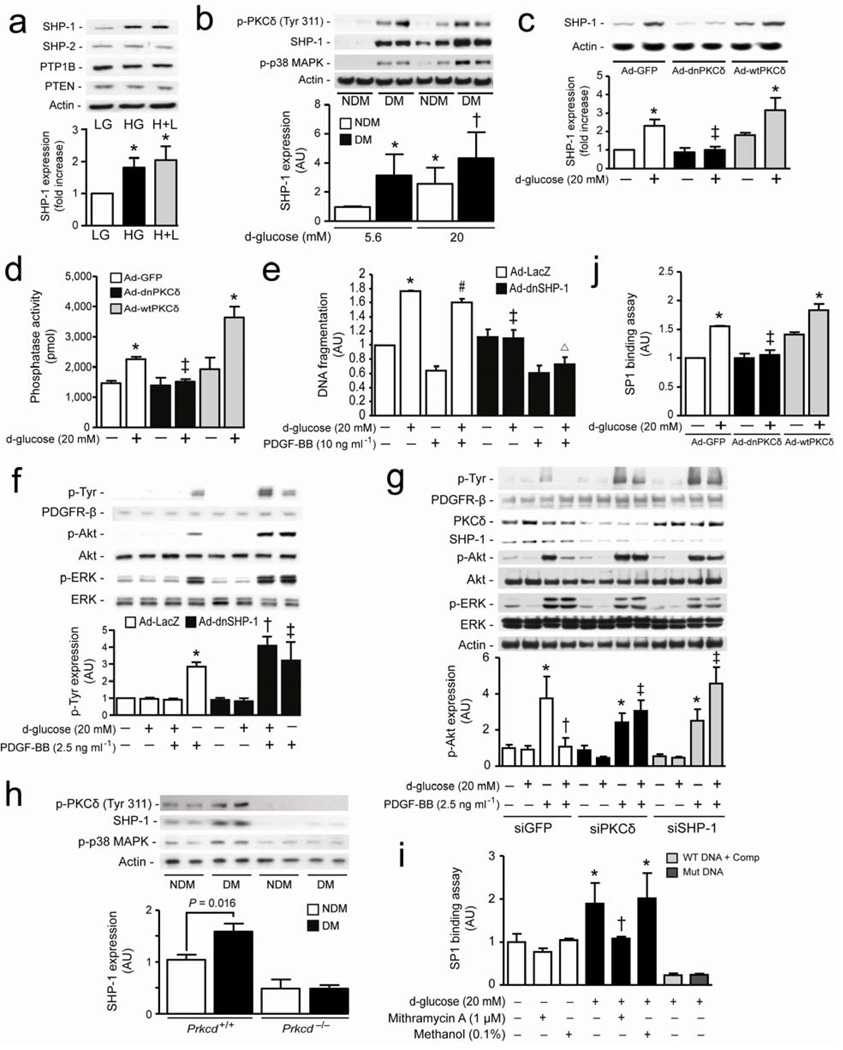
Cellular apoptosis induced by hyperglycemia occurs in many vascular cells and is crucial for the initiation of diabetic pathologies. In the retina, pericyte apoptosis and the formation of acellular capillaries, the most specific vascular pathologies attributed to hyperglycemia, is linked to the loss of platelet-derived growth factor (PDGF)-mediated survival actions owing to unknown mechanisms. Here we show that hyperglycemia persistently activates protein kinase C-delta (PKC-delta, encoded by Prkcd) and p38alpha mitogen-activated protein kinase (MAPK) to increase the expression of a previously unknown target of PKC-delta signaling, Src homology-2 domain-containing phosphatase-1 (SHP-1), a protein tyrosine phosphatase. This signaling cascade leads to PDGF receptor-beta dephosphorylation and a reduction in downstream signaling from this receptor, resulting in pericyte apoptosis independently of nuclear factor-kappaB (NF-kappaB) signaling. We observed increased PKC-delta activity and an increase in the number of acellular capillaries in diabetic mouse retinas, which were not reversible with insulin treatment that achieved normoglycemia. Unlike diabetic age-matched wild-type mice, diabetic Prkcd(-/-) mice did not show activation of p38alpha MAPK or SHP-1, inhibition of PDGF signaling in vascular cells or the presence of acellular capillaries. We also observed PKC-delta, p38alpha MAPK and SHP-1 activation in brain pericytes and in the renal cortex of diabetic mice. These findings elucidate a new signaling pathway by which hyperglycemia can induce PDGF resistance and increase vascular cell apoptosis to cause diabetic vascular complications.
Figures
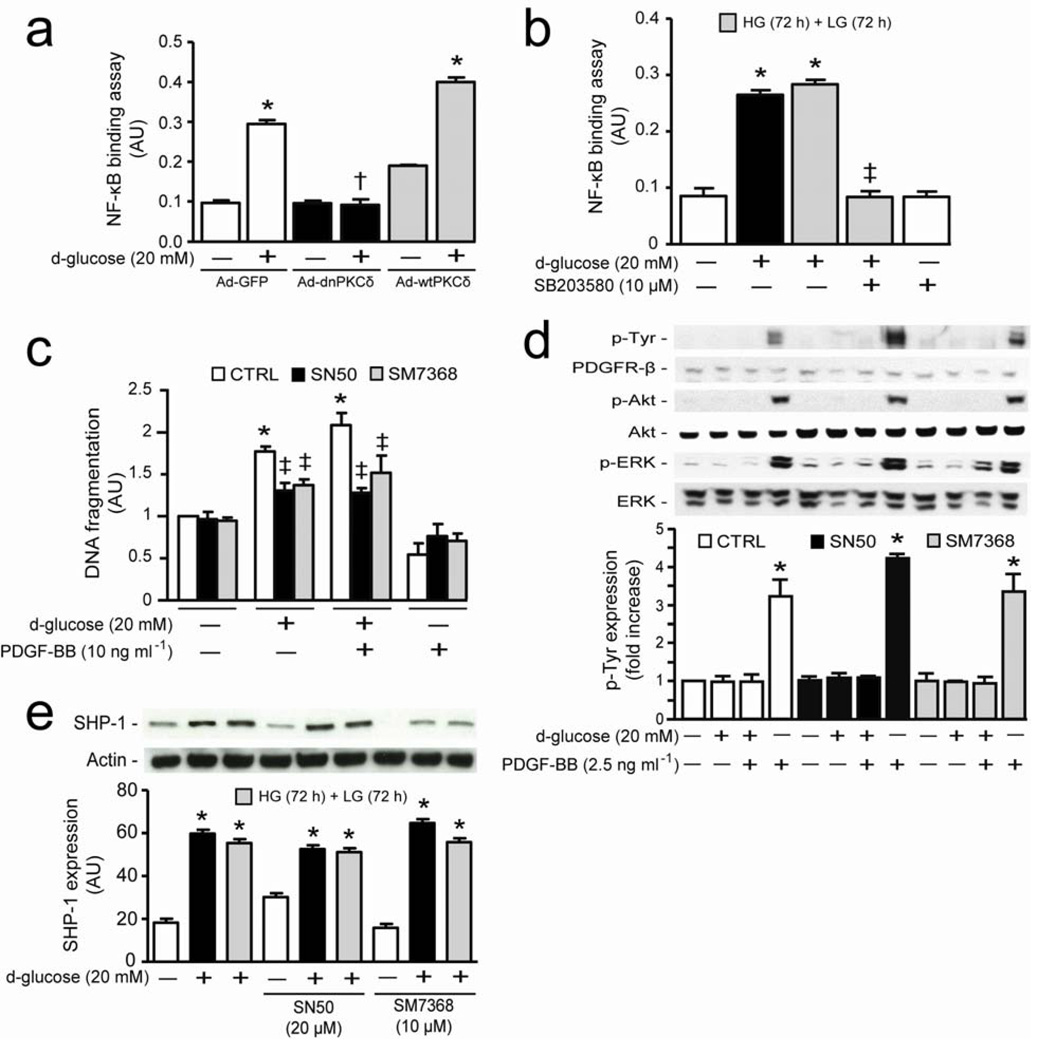
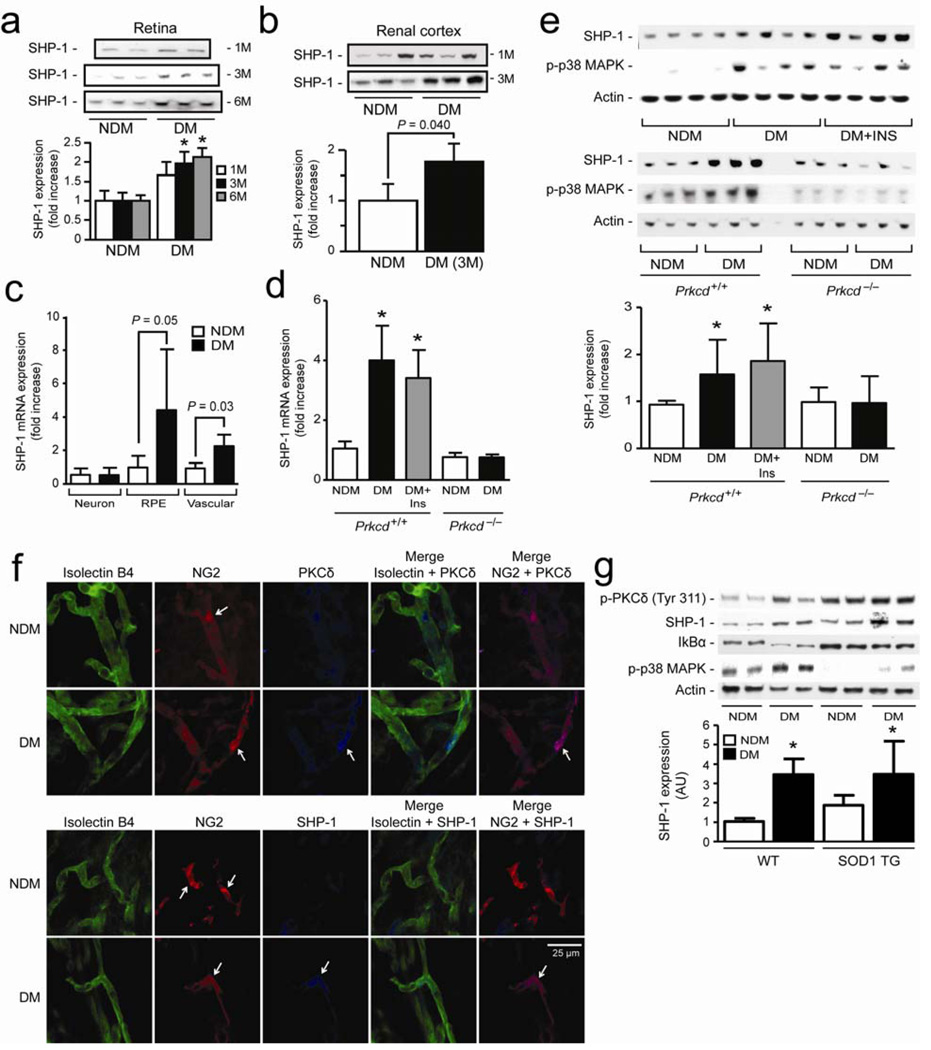
Comment in
-
Eye vessels saved by rescuing their pericyte partners.Nat Med. 2009 Nov;15(11):1248-9. doi: 10.1038/nm1109-1248. Nat Med. 2009. PMID: 19893554 No abstract available.
References
-
- Lorenzi M, Gerhardinger C. Early cellular and molecular changes induced by diabetes in the retina. Diabetologia. 2001;44:791–804. - PubMed
-
- Isermann B, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nature medicine. 2007;13:1349–1358. - PubMed
-
- >The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
