

Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice
- PMID: 19884661
- PMCID: PMC2786802
- DOI: 10.1172/JCI39845
Latent TGF-beta-binding protein 4 modifies muscular dystrophy in mice
Erratum in
- J Clin Invest. 2010 Feb;120(2):645
Abstract
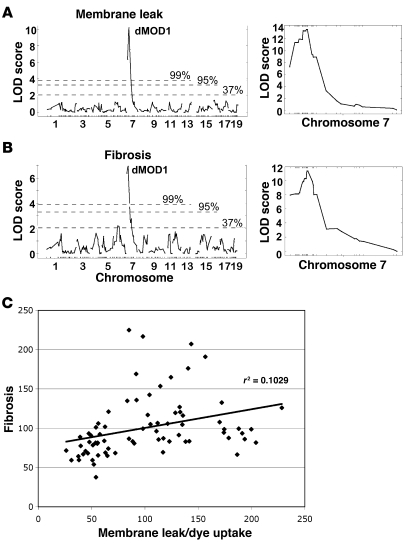
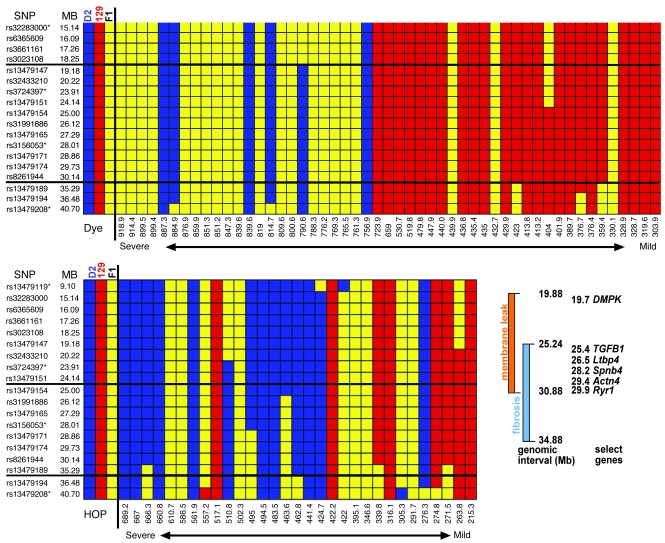
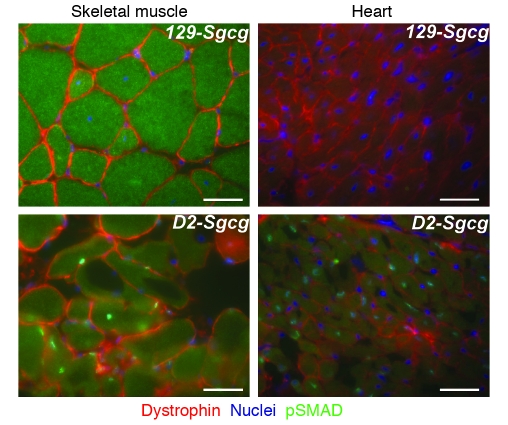
Most single-gene diseases, including muscular dystrophy, display a nonuniform phenotype. Phenotypic variability arises, in part, due to the presence of genetic modifiers that enhance or suppress the disease process. We employed an unbiased mapping approach to search for genes that modify muscular dystrophy in mice. In a genome-wide scan, we identified a single strong locus on chromosome 7 that influenced two pathological features of muscular dystrophy, muscle membrane permeability and muscle fibrosis. Within this genomic interval, an insertion/deletion polymorphism of 36 bp in the coding region of the latent TGF-beta-binding protein 4 gene (Ltbp4) was found. Ltbp4 encodes a latent TGF-beta-binding protein that sequesters TGF-beta and regulates its availability for binding to the TGF-beta receptor. Insertion of 12 amino acids into the proline-rich region of LTBP4 reduced proteolytic cleavage and was associated with reduced TGF-beta signaling, decreased fibrosis, and improved muscle pathology in a mouse model of muscular dystrophy. In contrast, a 12-amino-acid deletion in LTBP4 was associated with increased proteolysis, SMAD signaling, and fibrosis. These data identify Ltbp4 as a target gene to regulate TGF-beta signaling and modify outcomes in muscular dystrophy.
Figures





References
-
- Heydemann A., Doherty K.R., McNally E.M. Genetic modifiers of muscular dystrophy: implications for therapy. Biochim. Biophys. Acta. 2007;1772:216–228. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
