

Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression
- PMID: 19887574
- PMCID: PMC2792182
- DOI: 10.1101/gr.098921.109
Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression
Abstract
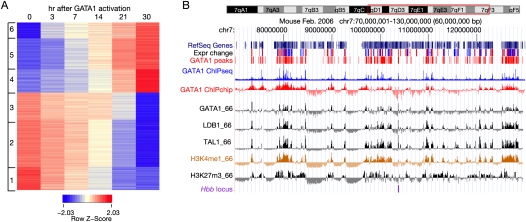
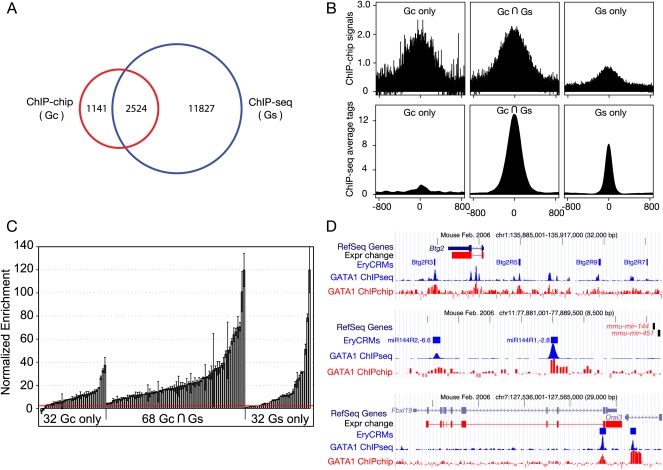
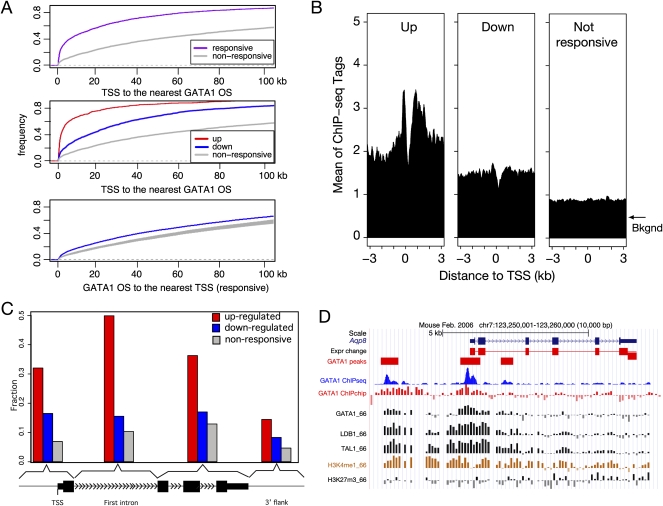
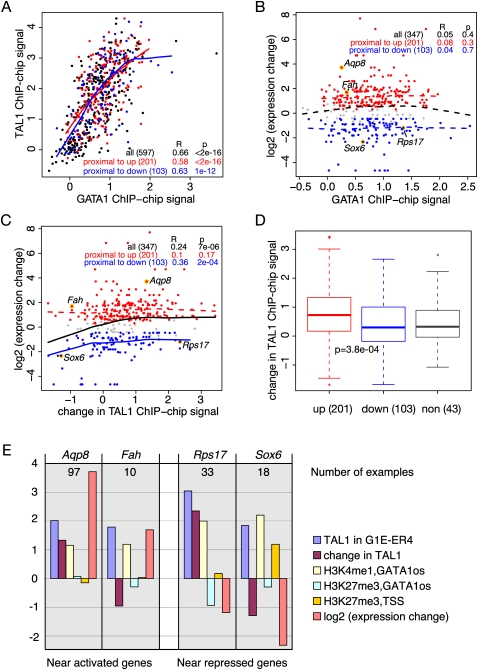
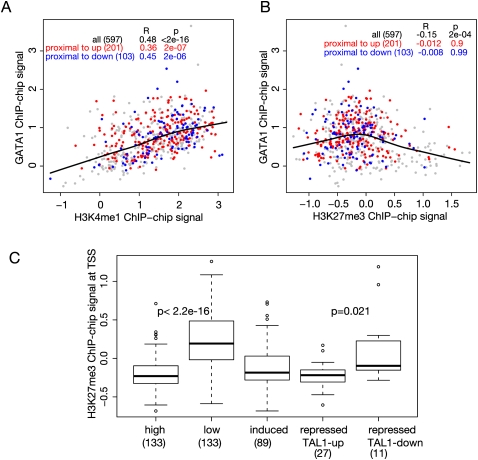
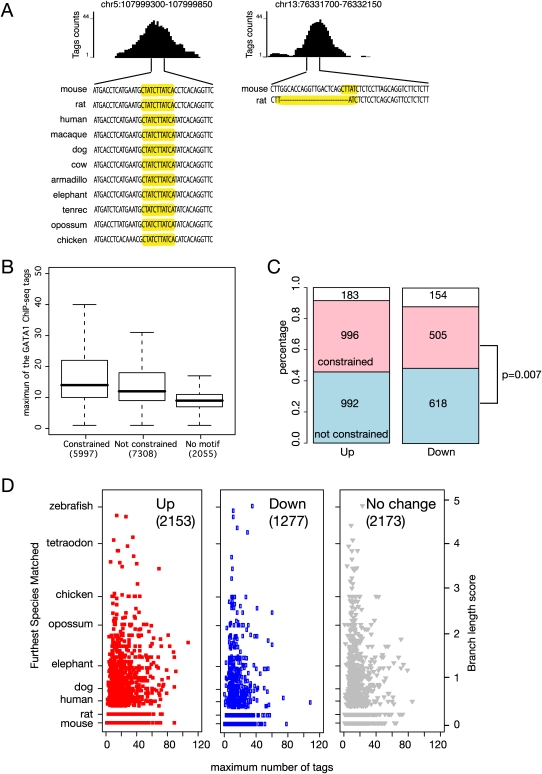
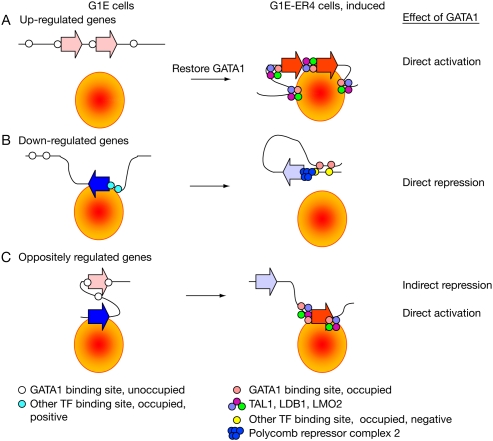
The transcription factor GATA1 regulates an extensive program of gene activation and repression during erythroid development. However, the associated mechanisms, including the contributions of distal versus proximal cis-regulatory modules, co-occupancy with other transcription factors, and the effects of histone modifications, are poorly understood. We studied these problems genome-wide in a Gata1 knockout erythroblast cell line that undergoes GATA1-dependent terminal maturation, identifying 2616 GATA1-responsive genes and 15,360 GATA1-occupied DNA segments after restoration of GATA1. Virtually all occupied DNA segments have high levels of H3K4 monomethylation and low levels of H3K27me3 around the canonical GATA binding motif, regardless of whether the nearby gene is induced or repressed. Induced genes tend to be bound by GATA1 close to the transcription start site (most frequently in the first intron), have multiple GATA1-occupied segments that are also bound by TAL1, and show evolutionary constraint on the GATA1-binding site motif. In contrast, repressed genes are further away from GATA1-occupied segments, and a subset shows reduced TAL1 occupancy and increased H3K27me3 at the transcription start site. Our data expand the repertoire of GATA1 action in erythropoiesis by defining a new cohort of target genes and determining the spatial distribution of cis-regulatory modules throughout the genome. In addition, we begin to establish functional criteria and mechanisms that distinguish GATA1 activation from repression at specific target genes. More broadly, these studies illustrate how a "master regulator" transcription factor coordinates tissue differentiation through a panoply of DNA and protein interactions.
Figures
References
-
- Blobel GA, Weiss MJ. Nuclear factors that regulate erythropoiesis. In: Steinberg MH, et al., editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge University Press; Cambridge, UK: 2001. pp. 72–94.
-
- Cantor A, Orkin S. Transcriptional regulation of erythropoiesis: An affair involving multiple partners. Oncogene. 2002;21:3368–3376. - PubMed
-
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases