

Edgetic perturbation models of human inherited disorders
- PMID: 19888216
- PMCID: PMC2795474
- DOI: 10.1038/msb.2009.80
Edgetic perturbation models of human inherited disorders
Abstract
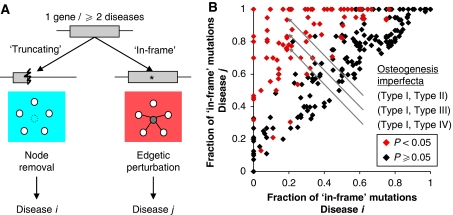
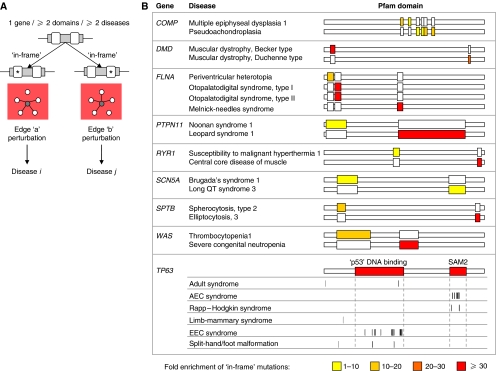
Cellular functions are mediated through complex systems of macromolecules and metabolites linked through biochemical and physical interactions, represented in interactome models as 'nodes' and 'edges', respectively. Better understanding of genotype-to-phenotype relationships in human disease will require modeling of how disease-causing mutations affect systems or interactome properties. Here we investigate how perturbations of interactome networks may differ between complete loss of gene products ('node removal') and interaction-specific or edge-specific ('edgetic') alterations. Global computational analyses of approximately 50,000 known causative mutations in human Mendelian disorders revealed clear separations of mutations probably corresponding to those of node removal versus edgetic perturbations. Experimental characterization of mutant alleles in various disorders identified diverse edgetic interaction profiles of mutant proteins, which correlated with distinct structural properties of disease proteins and disease mechanisms. Edgetic perturbations seem to confer distinct functional consequences from node removal because a large fraction of cases in which a single gene is linked to multiple disorders can be modeled by distinguishing edgetic network perturbations. Edgetic network perturbation models might improve both the understanding of dissemination of disease alleles in human populations and the development of molecular therapeutic strategies.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
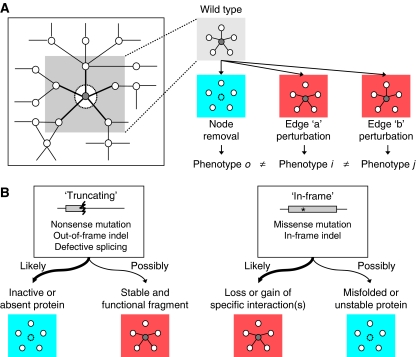
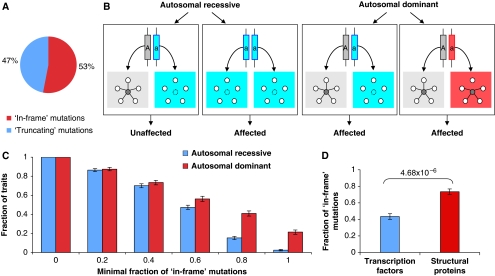
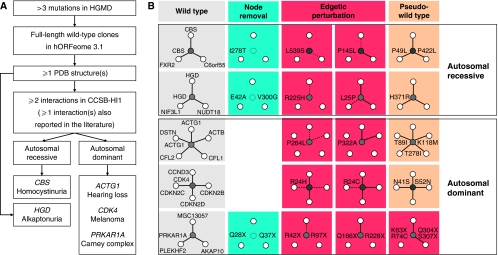
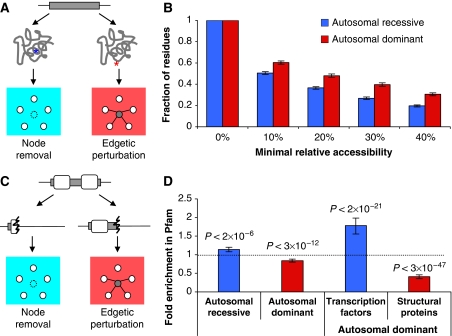
References
-
- Ball CA, Dolinski K, Dwight SS, Harris MA, Issel-Tarver L, Kasarskis A, Scafe CR, Sherlock G, Binkley G, Jin H, Kaloper M, Orr SD, Schroeder M, Weng S, Zhu Y, Botstein D, Cherry JM (2000) Integrating functional genomic information into the Saccharomyces genome database. Nucleic Acids Res 28: 77–80 - PMC - PubMed
-
- Botstein D, Risch N (2003) Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet 33 : 228–237 - PubMed
-
- Braun P, Tasan M, Dreze M, Barrios-Rodiles M, Lemmens I, Yu H, Sahalie JM, Murray RR, Roncari L, de Smet AS, Venkatesan K, Rual JF, Vandenhaute J, Cusick ME, Pawson T, Hill DE, Tavernier J, Wrana JL, Roth FP, Vidal M (2009) An experimentally derived confidence score for binary protein–protein interactions. Nat Methods 6: 91–97 - PMC - PubMed
-
- Chik JK, Lindberg U, Schutt CE (1996) The structure of an open state of β-actin at 2.65 Å resolution. J Mol Biol 263: 607–623 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
