

Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports
- PMID: 19889212
- PMCID: PMC2777856
- DOI: 10.1186/1750-1172-4-25
Recurrent microdeletion at 17q12 as a cause of Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: two case reports
Abstract
Background: Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH) consists of congenital aplasia of the uterus and the upper part of vagina due to anomalous development of Müllerian ducts, either isolated or associated with other congenital malformations, including renal, skeletal, hearing and heart defects. This disorder has an incidence of approximately 1 in 4500 newborn girls and the aetiology is poorly understood.
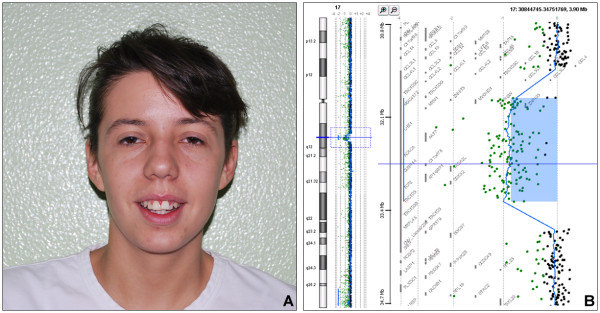
Methods and results: we report on two patients affected by MRKH syndrome in which array-CGH analysis disclosed an identical deletion spanning 1.5 Mb of genomic DNA at chromosome 17q12. One patient was affected by complete absence of uterus and vagina, with bilaterally normal ovaries, while the other displayed agenesis of the upper part of vagina, right unicornuate uterus, non cavitating rudimentary left horn and bilaterally multicystic kidneys. The deletion encompassed two candidate genes, TCF2 and LHX1. Mutational screening of these genes in a selected group of 20 MRKH females without 17q12 deletion was negative.
Conclusion: Deletion 17q12 is a rare albeit recurrent anomaly mediated by segmental duplications, previously reported in subjects with developmental kidney abnormalities and diabetes. The present two patients expand the clinical spectrum associated with this imbalance and suggest that this region is a candidate locus for a subset of MRKH syndrome individuals, with or without renal defects.
Figures
References
-
- Pittock ST, Babovic-Vuksanovic D, Lteif A. Mayer-Rokitansky-Kuster-Hauser anomaly and its associated malformations. Am J Med Genet A. 2005;135:314–316. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
