

Interaction of staphylococci with bone
- PMID: 19889575
- PMCID: PMC2814006
- DOI: 10.1016/j.ijmm.2009.10.003
Interaction of staphylococci with bone
Abstract
Staphylococci, in particular Staphylococcus aureus, are the predominant cause of bone infections worldwide. These infections are painful, debilitating and with the rise in antibiotic-resistant forms, increasingly difficult to treat. The growth in the number of prosthetic joint replacement procedures also provides new opportunities for these infections to take hold. Comprehending the mechanisms by which staphylococci interact with and damage bone is critical to the development of new approaches to meet this challenge. This review summarises current understanding of the mechanisms by which staphylococci infect and damage bone. We address the role of the inflammatory response to staphylococcal infection in disrupting the homeostatic balance of bone matrix deposition and resorption and thereby mediating bone destruction. A number of virulence factors that have been shown to contribute to bone infection and pathology are discussed, however no single factor has been defined as being specific to bone infections. Although traditionally considered an extracellular pathogen, there is increasing evidence that staphylococci are able to invade host cells, and that an intracellular lifestyle may facilitate long-term persistence in bone tissue, enabling evasion of antimicrobials and host immune responses. 'Small colony variant' strains, with mutations disabling the electron transport pathway appear particularly adept at invading and persisting within host cells, and exhibit enhanced antimicrobial resistance, and may represent a further complication in the treatment and management of staphylococcal bone disease.
Copyright 2009 Elsevier GmbH. All rights reserved.
Figures
References
-
- Abdelnour A., Bremell T., Tarkowski A. Toxic shock syndrome toxin 1 contributes to the arthritogenicity of Staphylococcus aureus. J. Infect. Dis. 1994;170:94–99. - PubMed
-
- Adler H., Widmer A., Frei R. Emergence of a teicoplanin-resistant small colony variant of Staphylococcus epidermidis during vancomycin therapy. Eur. J. Clin. Microbiol. Infect. Dis. 2003;22:746–748. - PubMed
-
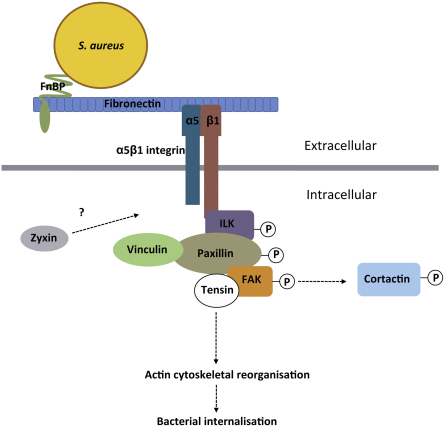
- Agerer F., Lux S., Michel A., Rohde M., Ohlsen K., Hauck C.R. Cellular invasion by Staphylococcus aureus reveals a functional link between focal adhesion kinase and cortactin in integrin-mediated internalisation. J. Cell Sci. 2005;118:2189–2200. - PubMed
-
- Aggarwal B.B. Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 2003;3:745–756. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
