

Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer's disease
- PMID: 19890241
- PMCID: PMC3056539
- DOI: 10.1017/s1092852900024901
Role of mitochondria in neurodegenerative diseases: mitochondria as a therapeutic target in Alzheimer's disease
Abstract
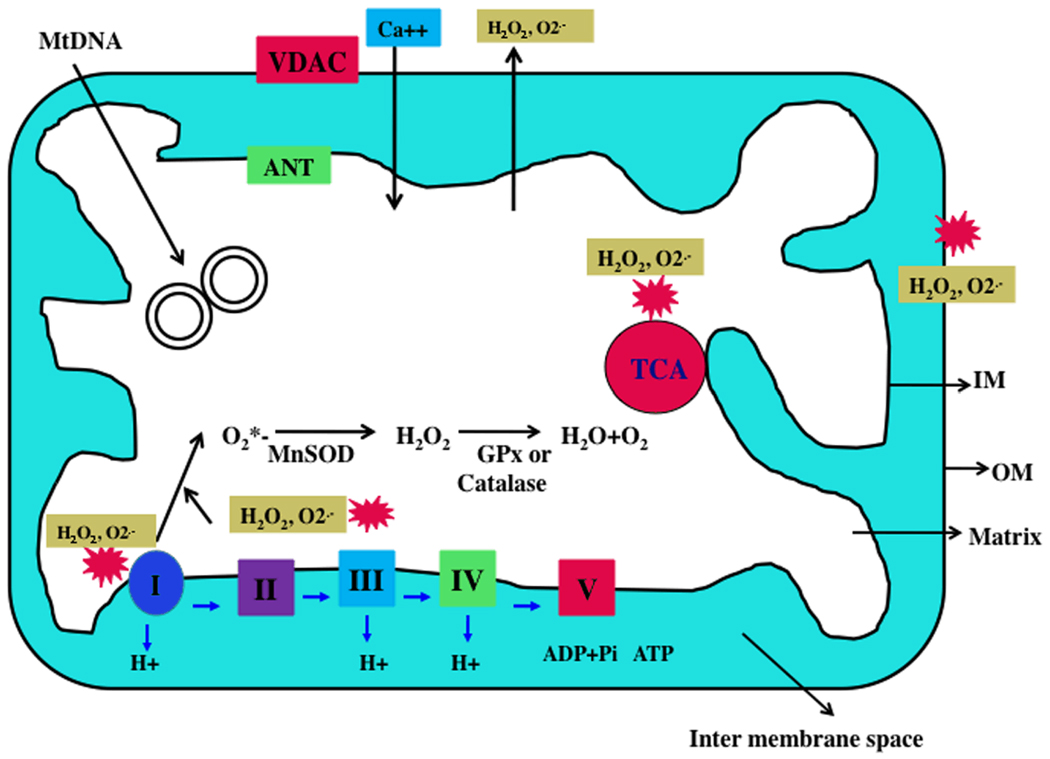
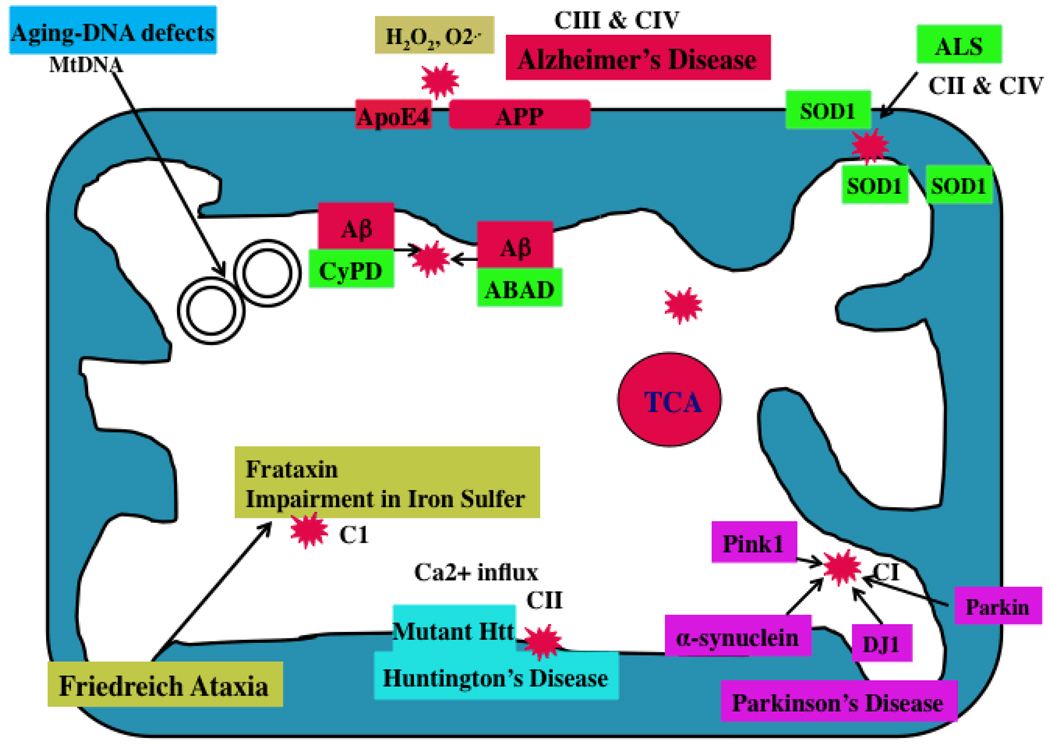
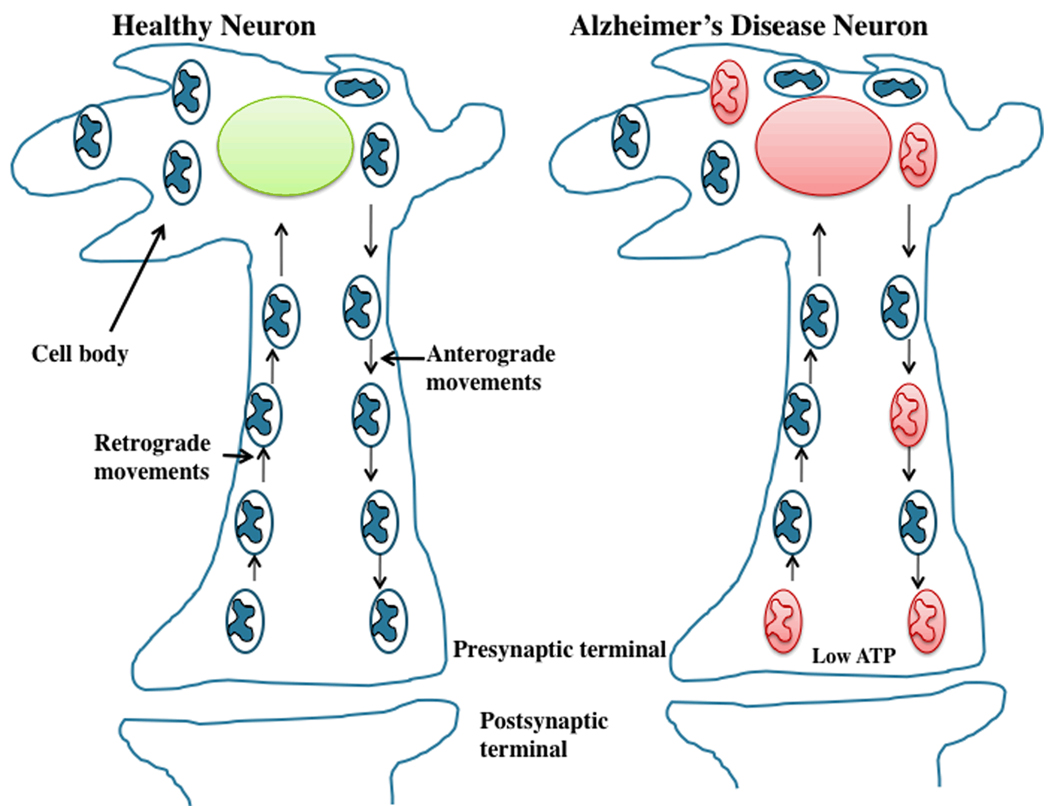
A growing body of evidence suggests that mitochondrial abnormalities are involved in aging and in age-related neurodegenerative diseases as well as cancer, diabetes, and several other diseases known to be affected by mitochondria. Causal factors for most age-related neurodegenerative diseases-including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis (ALS), and Friedrich ataxia (FRDA)-are largely unknown. Genetic defects are reported to cause a small number of neurodegenerative diseases, but cellular, molecular, and pathological mechanisms of disease progression and selective neuronal cell death are not understood fully in these diseases. However, based on several cellular, molecular, and animal model studies of Alzheimer's disease, Parkinson's disease, ALS, FRDA, cancer, and diabetes, aging may play a large role in cell death in these diseases. Age-dependent, mitochondrially-generated reactive oxygen species (ROS) have been identified as important factors responsible for disease progression and cell death, particularly in late-onset diseases, in which genetic mutations are not causal factors.
Figures
References
-
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. - PubMed
-
- Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58:495–505. - PubMed
-
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. - PubMed
-
- Chandrasekaran K, Giordano T, Brady DR, Stoll J, Martin LJ, Rapoport SI. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res Mol Brain Res. 1994;24:336–340. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous