

Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter
- PMID: 19892699
- PMCID: PMC2804362
- DOI: 10.1074/jbc.M109.072587
Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter
Abstract
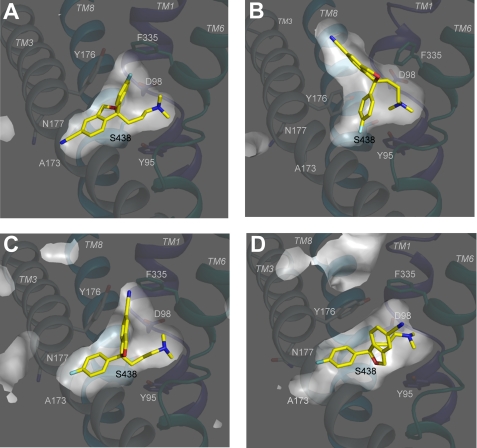
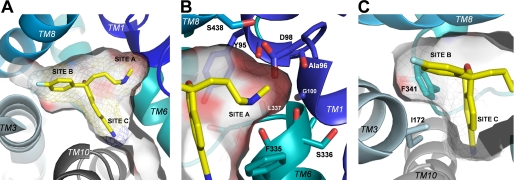
The serotonin transporter (SERT) regulates extracellular levels of the neurotransmitter serotonin (5-hydroxytryptamine) in the brain by facilitating uptake of released 5-hydroxytryptamine into neuronal cells. SERT is the target for widely used antidepressant drugs, including imipramine, fluoxetine, and (S)-citalopram, which are competitive inhibitors of the transport function. Knowledge of the molecular details of the antidepressant binding sites in SERT has been limited due to lack of structural data on SERT. Here, we present a characterization of the (S)-citalopram binding pocket in human SERT (hSERT) using mutational and computational approaches. Comparative modeling and ligand docking reveal that (S)-citalopram fits into the hSERT substrate binding pocket, where (S)-citalopram can adopt a number of different binding orientations. We find, however, that only one of these binding modes is functionally relevant from studying the effects of 64 point mutations around the putative substrate binding site. The mutational mapping also identify novel hSERT residues that are crucial for (S)-citalopram binding. The model defines the molecular determinants for (S)-citalopram binding to hSERT and demonstrates that the antidepressant binding site overlaps with the substrate binding site.
Figures
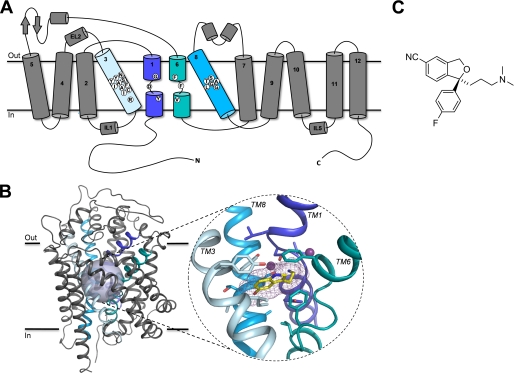

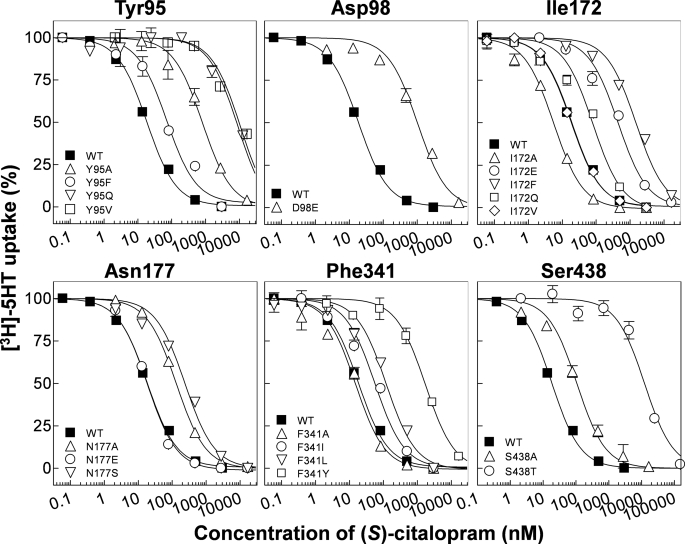
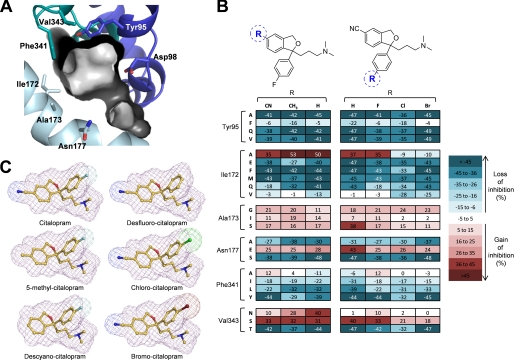
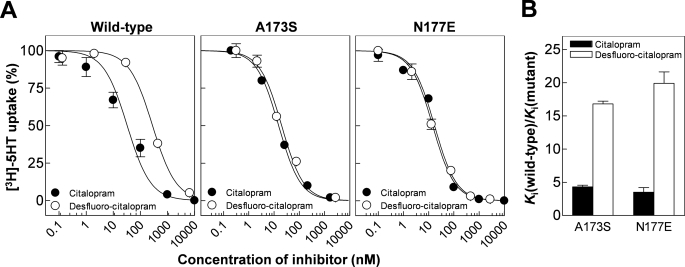
References
-
- Masson J., Sagné C., Hamon M., El Mestikawy S. (1999) Pharmacol. Rev. 51, 439–464 - PubMed
-
- Gether U., Andersen P. H., Larsson O. M., Schousboe A. (2006) Trends Pharmacol. Sci. 27, 375–383 - PubMed
-
- Rudnick G. (2006) J. Membr. Biol. 213, 101–110 - PubMed
-
- Chen N. H., Reith M. E., Quick M. W. (2004) Pflugers Arch. 447, 519–531 - PubMed
-
- Moltzen E. K., Bang-Andersen B. (2006) Curr. Top. Med. Chem. 6, 1801–1823 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
