

CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila
- PMID: 19896112
- PMCID: PMC2775834
- DOI: 10.1016/j.ajhg.2009.10.004
CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila
Abstract
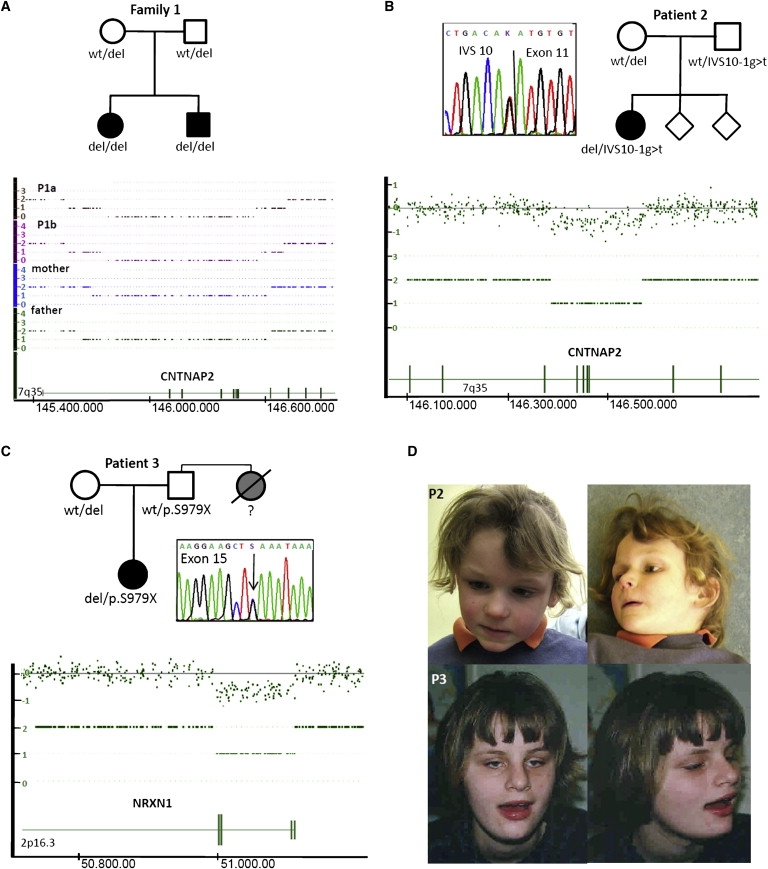
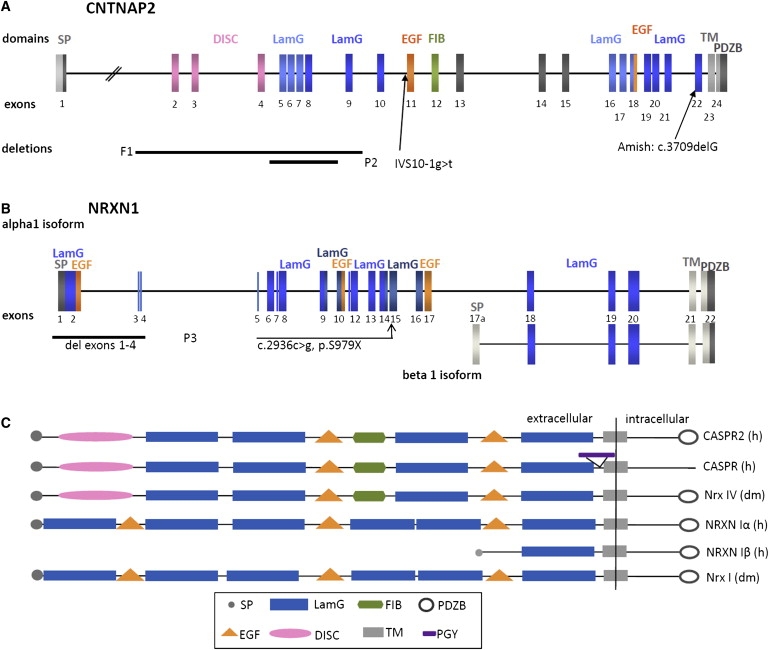
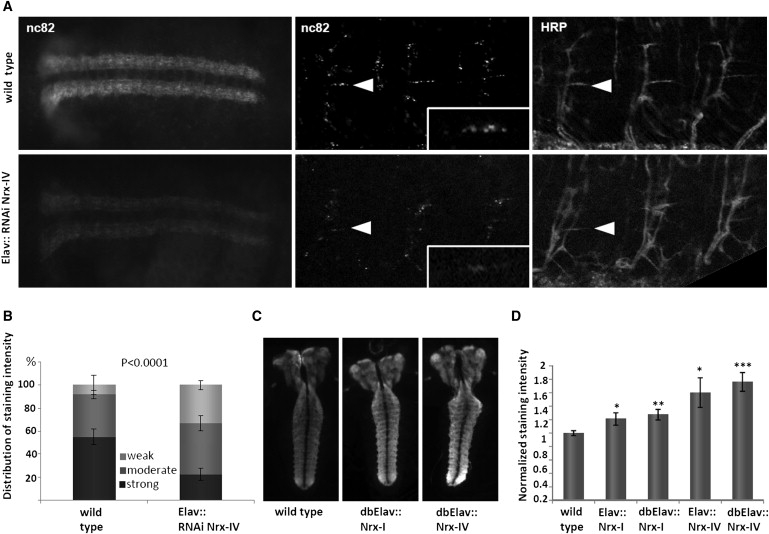
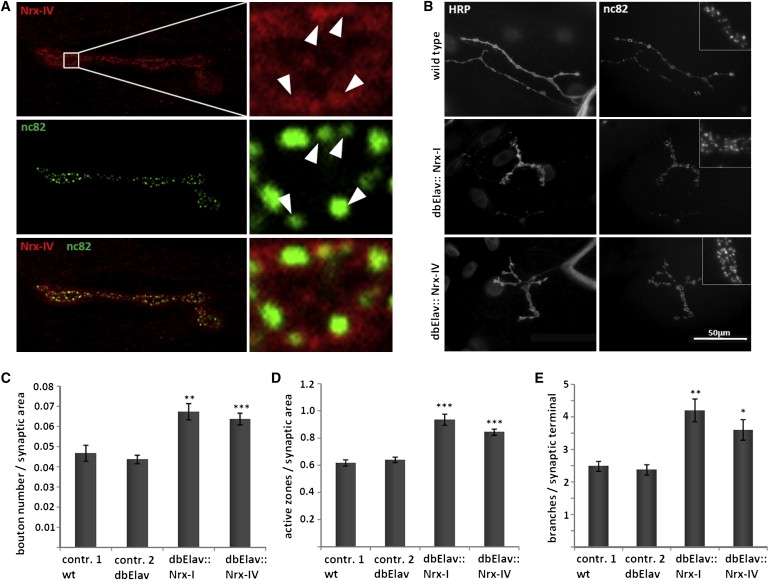
Heterozygous copy-number variants and SNPs of CNTNAP2 and NRXN1, two distantly related members of the neurexin superfamily, have been repeatedly associated with a wide spectrum of neuropsychiatric disorders, such as developmental language disorders, autism spectrum disorders, epilepsy, and schizophrenia. We now identified homozygous and compound-heterozygous deletions and mutations via molecular karyotyping and mutational screening in CNTNAP2 and NRXN1 in four patients with severe mental retardation (MR) and variable features, such as autistic behavior, epilepsy, and breathing anomalies, phenotypically overlapping with Pitt-Hopkins syndrome. With a frequency of at least 1% in our cohort of 179 patients, recessive defects in CNTNAP2 appear to significantly contribute to severe MR. Whereas the established synaptic role of NRXN1 suggests that synaptic defects contribute to the associated neuropsychiatric disorders and to severe MR as reported here, evidence for a synaptic role of the CNTNAP2-encoded protein CASPR2 has so far been lacking. Using Drosophila as a model, we now show that, as known for fly Nrx-I, the CASPR2 ortholog Nrx-IV might also localize to synapses. Overexpression of either protein can reorganize synaptic morphology and induce increased density of active zones, the synaptic domains of neurotransmitter release. Moreover, both Nrx-I and Nrx-IV determine the level of the presynaptic active-zone protein bruchpilot, indicating a possible common molecular mechanism in Nrx-I and Nrx-IV mutant conditions. We therefore propose that an analogous shared synaptic mechanism contributes to the similar clinical phenotypes resulting from defects in human NRXN1 and CNTNAP2.
Figures
References
-
- Rauch A., Hoyer J., Guth S., Zweier C., Kraus C., Becker C., Zenker M., Huffmeier U., Thiel C., Ruschendorf F. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. A. 2006;140:2063–2074. - PubMed
-
- Humeau Y., Gambino F., Chelly J., Vitale N. X-linked mental retardation: focus on synaptic function and plasticity. J. Neurochem. 2009;109:1–14. - PubMed
-
- Vaillend C., Poirier R., Laroche S. Genes, plasticity and mental retardation. Behav. Brain Res. 2008;192:88–105. - PubMed
-
- Amiel J., Rio M., de Pontual. L., Redon R., Malan V., Boddaert N., Plouin P., Carter N.P., Lyonnet S., Munnich A. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am. J. Hum. Genet. 2007;80:988–993. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
