

TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness
- PMID: 19896113
- PMCID: PMC2775830
- DOI: 10.1016/j.ajhg.2009.10.013
TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness
Abstract
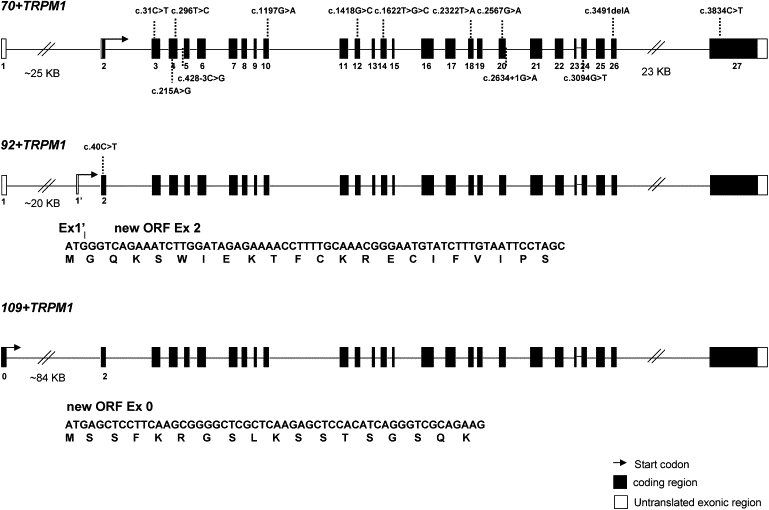
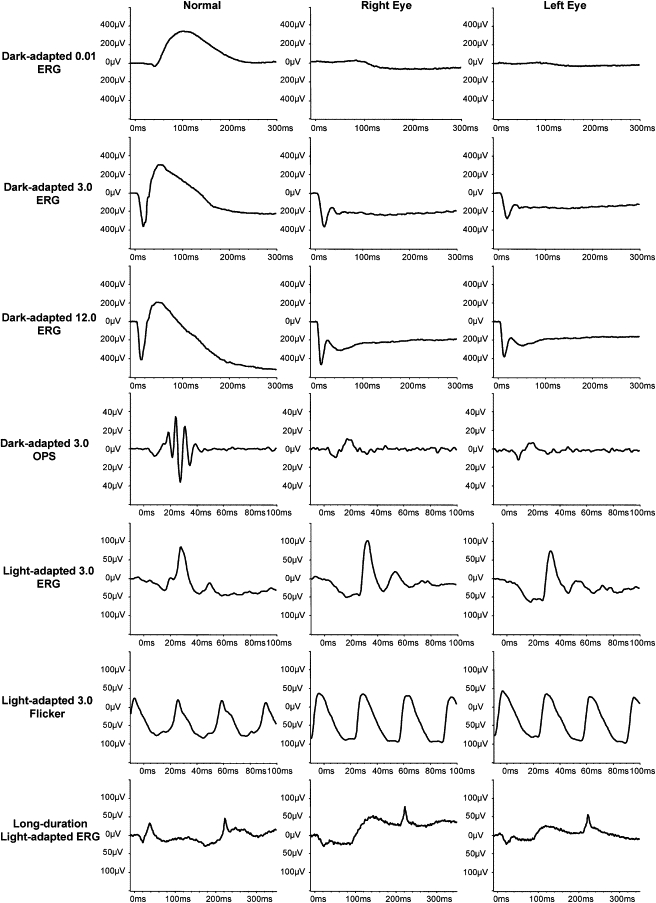
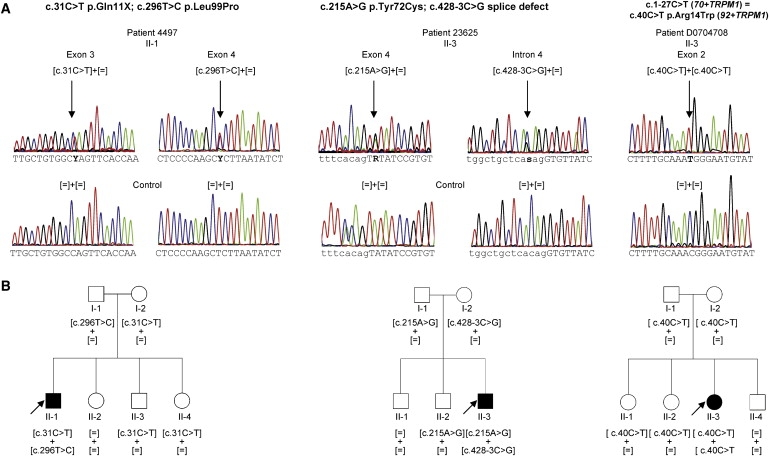
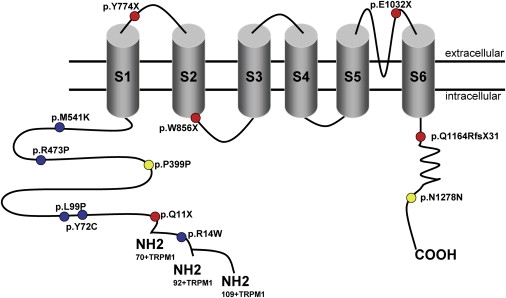
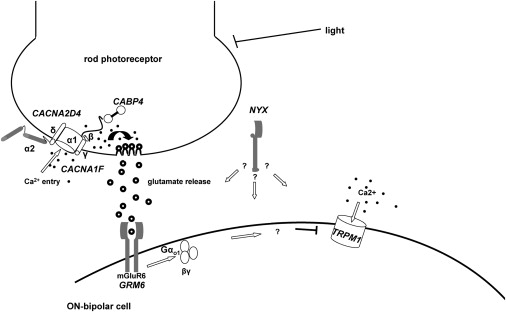
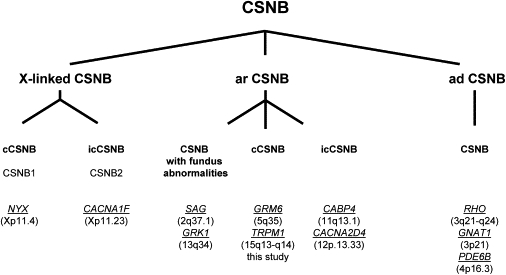
Night vision requires signaling from rod photoreceptors to adjacent bipolar cells in the retina. Mutations in the genes NYX and GRM6, expressed in ON bipolar cells, lead to a disruption of the ON bipolar cell response. This dysfunction is present in patients with complete X-linked and autosomal-recessive congenital stationary night blindness (CSNB) and can be assessed by standard full-field electroretinography (ERG), showing severely reduced rod b-wave amplitude and slightly altered cone responses. Although many cases of complete CSNB (cCSNB) are caused by mutations in NYX and GRM6, in approximately 60% of the patients the gene defect remains unknown. Animal models of human diseases are a good source for candidate genes, and we noted that a cCSNB phenotype present in homozygous Appaloosa horses is associated with downregulation of TRPM1. TRPM1, belonging to the family of transient receptor potential channels, is expressed in ON bipolar cells and therefore qualifies as an excellent candidate. Indeed, mutation analysis of 38 patients with CSNB identified ten unrelated cCSNB patients with 14 different mutations in this gene. The mutation spectrum comprises missense, splice-site, deletion, and nonsense mutations. We propose that the cCSNB phenotype in these patients is due to the absence of functional TRPM1 in retinal ON bipolar cells.
Figures

References
-
- Zeitz C. Molecular genetics and protein function involved in nocturnal vision. Expert Rev Ophthalmol. 2007;2:467–485.
-
- Schubert G., Bornschein H. Ophthalmologica. 1952;123:396–413. - PubMed
-
- Miyake Y., Yagasaki K., Horiguchi M., Kawase Y., Kanda T. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch. Ophthalmol. 1986;104:1013–1020. - PubMed
-
- Audo I., Robson A.G., Holder G.E., Moore A.T. The negative ERG: clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv. Ophthalmol. 2008;53:16–40. - PubMed
-
- Zeitz C., Labs S., Lorenz B., Forster U., Ueksti J., Kroes H.Y., De Baere E., Leroy B.P., Cremers F.P., Wittmer M. Genotyping microarray for CSNB-associated genes. Invest Ophthalmol Vis Sci. 2009 Published online July 2, 2009. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
