

Comparative transcriptional and genomic analysis of Plasmodium falciparum field isolates
- PMID: 19898609
- PMCID: PMC2764095
- DOI: 10.1371/journal.ppat.1000644
Comparative transcriptional and genomic analysis of Plasmodium falciparum field isolates
Abstract
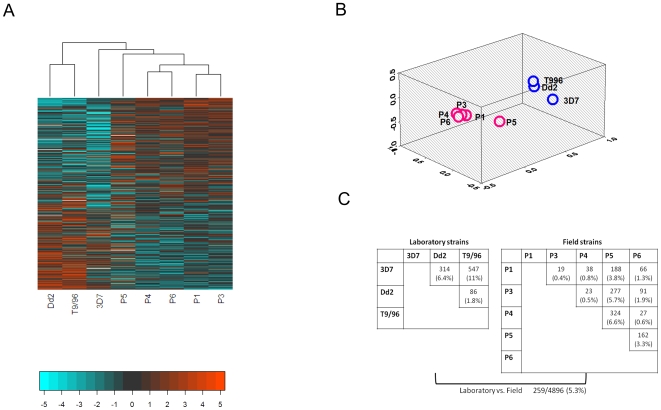
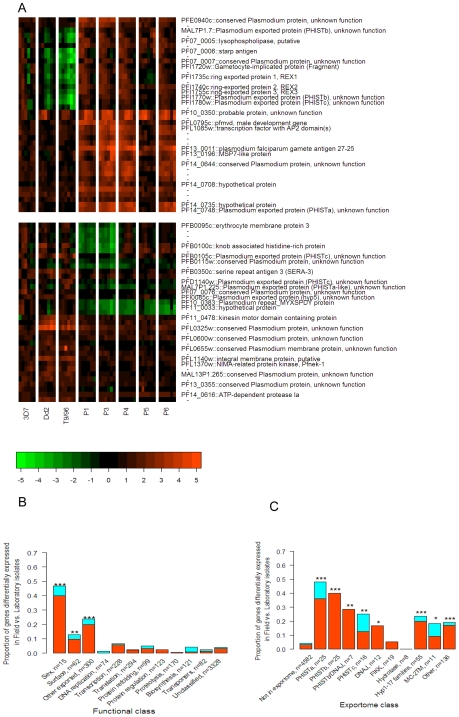
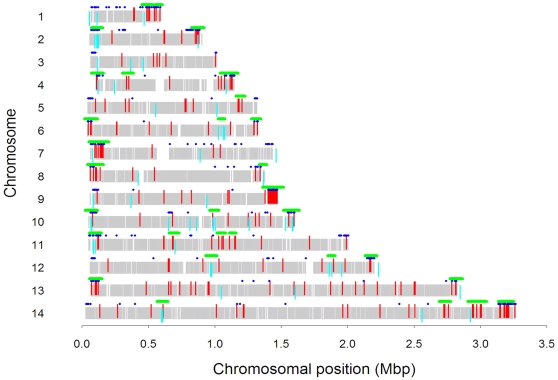
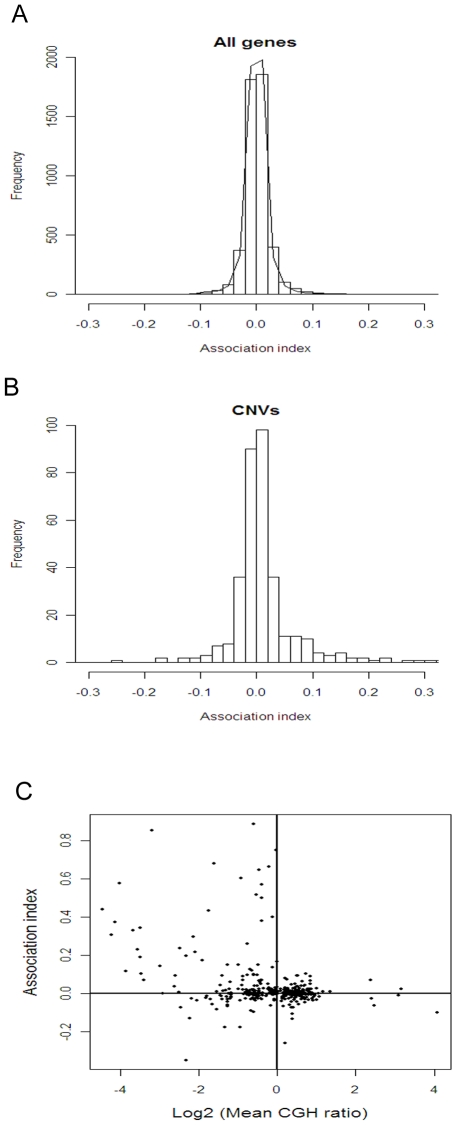
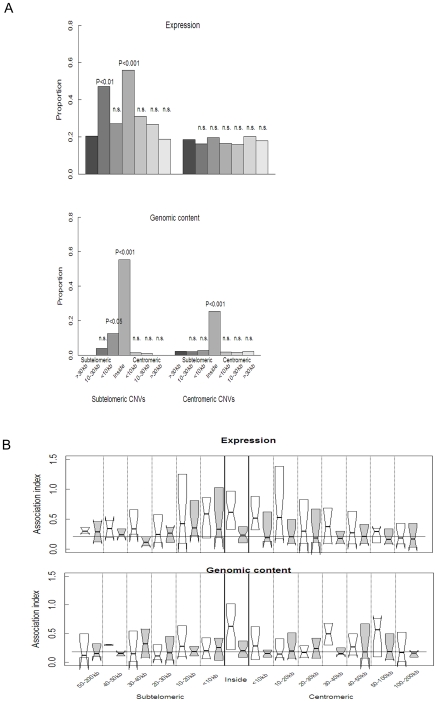
Mechanisms for differential regulation of gene expression may underlie much of the phenotypic variation and adaptability of malaria parasites. Here we describe transcriptional variation among culture-adapted field isolates of Plasmodium falciparum, the species responsible for most malarial disease. It was found that genes coding for parasite protein export into the red cell cytosol and onto its surface, and genes coding for sexual stage proteins involved in parasite transmission are up-regulated in field isolates compared with long-term laboratory isolates. Much of this variability was associated with the loss of small or large chromosomal segments, or other forms of gene copy number variation that are prevalent in the P. falciparum genome (copy number variants, CNVs). Expression levels of genes inside these segments were correlated to that of genes outside and adjacent to the segment boundaries, and this association declined with distance from the CNV boundary. This observation could not be explained by copy number variation in these adjacent genes. This suggests a local-acting regulatory role for CNVs in transcription of neighboring genes and helps explain the chromosomal clustering that we observed here. Transcriptional co-regulation of physical clusters of adaptive genes may provide a way for the parasite to readily adapt to its highly heterogeneous and strongly selective environment.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
