

Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death
- PMID: 19901028
- PMCID: PMC2804228
- DOI: 10.1074/jbc.M109.070037
Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death
Abstract
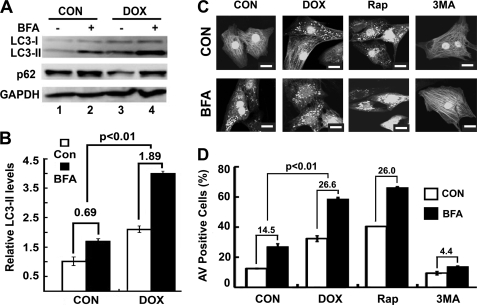
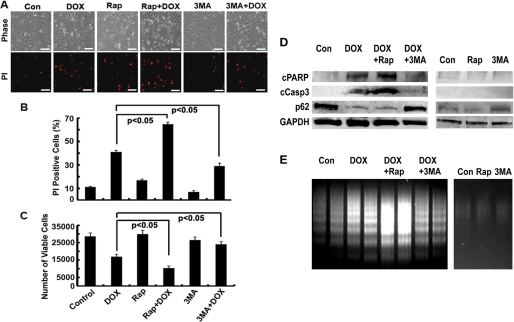
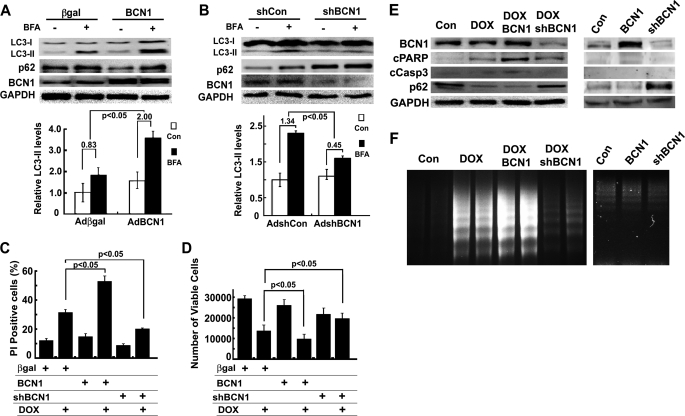
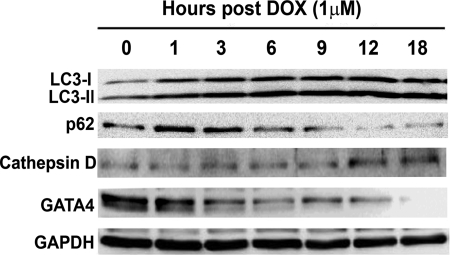
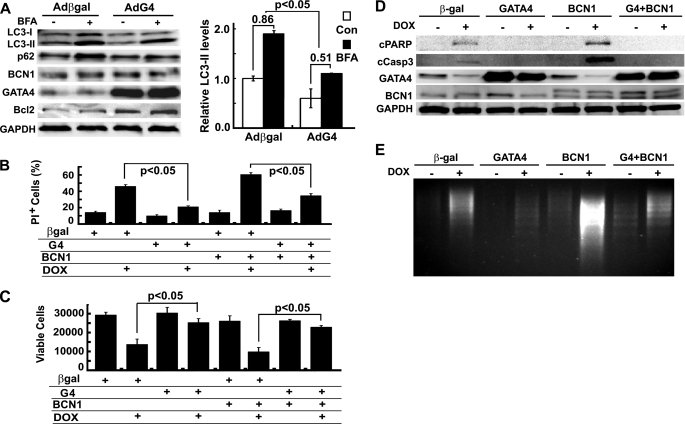
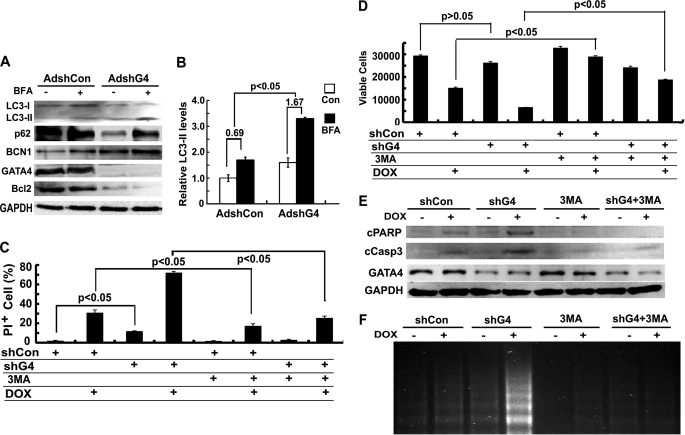
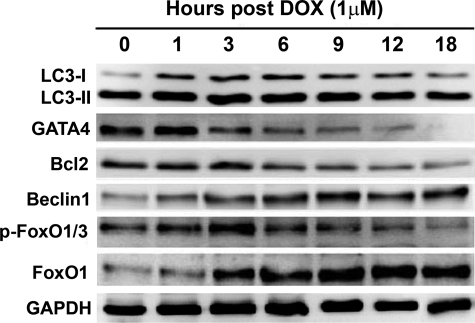
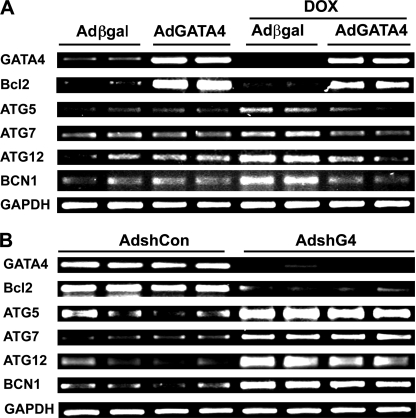
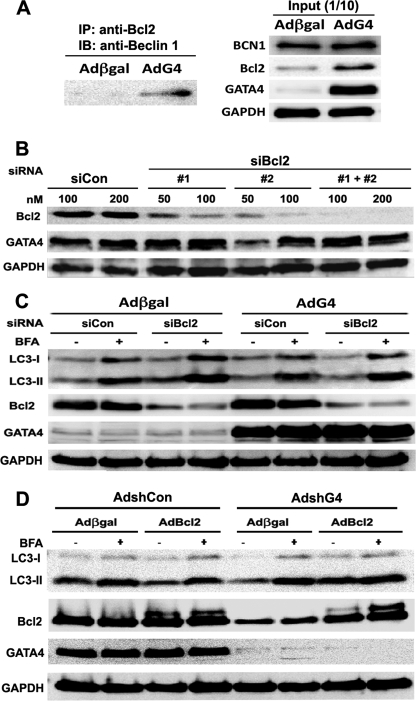
Doxorubicin (DOX) is a potent anti-tumor drug known to cause heart failure. The transcription factor GATA4 antagonizes DOX-induced cardiotoxicity. However, the protective mechanism remains obscure. Autophagy is the primary cellular pathway for lysosomal degradation of long-lived proteins and organelles, and its activation could be either protective or detrimental depending on specific pathophysiological conditions. Here we investigated the ability of GATA4 to inhibit autophagy as a potential mechanism underlying its protection against DOX toxicity in cultured neonatal rat cardiomyocytes. DOX markedly increased autophagic flux in cardiomyocytes as indicated by the difference in protein levels of LC3-II (microtubule-associated protein light chain 3 form 2) or numbers of autophagic vacuoles in the absence and presence of the lysosomal inhibitor bafilomycin A1. DOX-induced cardiomyocyte death determined by multiple assays was aggravated by a drug or genetic approach that activates autophagy, but it was attenuated by manipulations that inhibit autophagy, suggesting that autophagy contributes to DOX cardiotoxicity. DOX treatment depleted GATA4 protein levels, which predisposed cardiomyocytes to DOX toxicity. Indeed, GATA4 gene silencing triggered autophagy that rendered DOX more toxic, whereas GATA4 overexpression inhibited DOX-induced autophagy, reducing cardiomyocyte death. Mechanistically, GATA4 up-regulated gene expression of the survival factor Bcl2 and suppressed DOX-induced activation of autophagy-related genes, which may likely be responsible for the anti-apoptotic and anti-autophagic effects of GATA4. Together, these findings suggest that activation of autophagy mediates DOX cardiotoxicity, and preservation of GATA4 attenuates DOX cardiotoxicity by inhibiting autophagy through modulation of the expression of Bcl2 and autophagy-related genes.
Figures
References
-
- Swain S. M., Whaley F. S., Ewer M. S. (2003) Cancer 97, 2869–2879 - PubMed
-
- Singal P. K., Iliskovic N. (1998) N. Engl. J. Med. 339, 900–905 - PubMed
-
- Minotti G., Menna P., Salvatorelli E., Cairo G., Gianni L. (2004) Pharmacol. Rev. 56, 185–229 - PubMed
-
- Kumar D., Kirshenbaum L. A., Li T., Danelisen I., Singal P. K. (2001) Antioxid. Redox Signal. 3, 135–145 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
