

Cellular prion protein mediates the toxicity of beta-amyloid oligomers: implications for Alzheimer disease
- PMID: 19901162
- PMCID: PMC2849161
- DOI: 10.1001/archneurol.2009.223
Cellular prion protein mediates the toxicity of beta-amyloid oligomers: implications for Alzheimer disease
Abstract
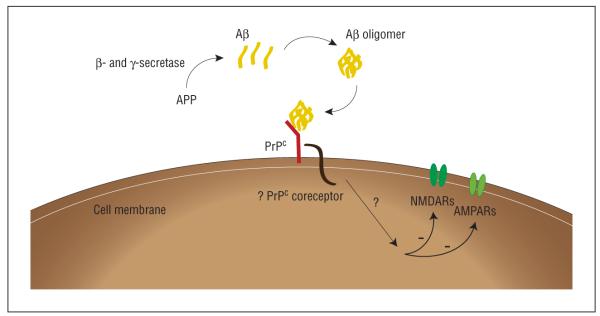
Alzheimer disease (AD) is the most common cause of age-related dementia, affecting more than 25 million people worldwide. The accumulation of insoluble beta-amyloid (Abeta) plaques in the brain has long been considered central to the pathogenesis of AD. However, recent evidence suggests that soluble oligomeric assemblies of Abeta may be of greater importance. beta-Amyloid oligomers have been found to be potent synaptotoxins, but the mechanism by which they exert their action has remained elusive. Herein, we review the recently published finding that cellular prion protein (PrP(c)) is a high-affinity receptor for Abeta oligomers, mediating their toxic effects on synaptic plasticity. We further discuss the relationship between AD and PrP(c) and the potential clinical implications. Cellular prion protein may provide a novel target for therapeutic intervention in AD.
Figures
References
-
- Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3(3):186–191. - PubMed
-
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. [published correction appears in Science. 2002;297(5590):2209] - PubMed
-
- Younkin SG. The role of Aβ42 in Alzheimer’s disease. J Physiol Paris. 1998;92(34):289–292. - PubMed
-
- Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
